

Abstract
Potential energy surfaces for reactions involving NH
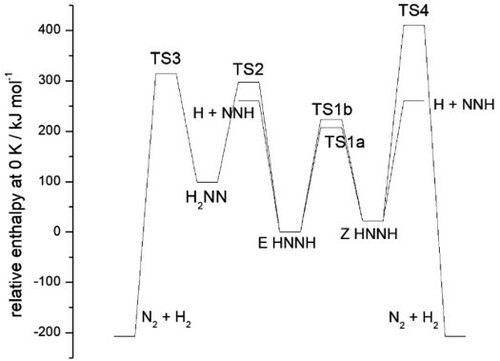
isomers of diazene (diimide) have been explored using density functional theory, with energies based on coupled-cluster theory. A focus is on processes that create or consume these species, and isomerisation between the E (trans) and Z (cis) forms of HNNH. These include isomerisation and dissociation pathways for HNNH, addition of H atoms to form N
H
, abstraction by H atoms yielding short-lived NNH, and abstraction reactions of H with N
H
. Transition state and capture theories are applied for high-pressure-limiting behaviour, while low-pressure and falloff regions are characterised via the methods of Troe and coworkers. Rate constants and thermochemistry are provided to improve models of diamine chemistry, relevant to the combustion of NH
especially at high concentrations, high pressures or under reducing conditions. Results indicate that amine radical recombination mainly yields the E HNNH isomer, while H-abstraction from N
H
results in E HNNH and H
NN. However, at elevated temperature E
Z isomerisation becomes competitive, and Z HNNH, being more reactive, acts to enhance the diazene consumption rate.
GRAPHICAL ABSTRACT
Acknowledgments
PM thanks the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences and Biosciences/Gas Phase Chemical Physics under Contract No. DESC0020952. Computational facilities were provided by the National Science Foundation, Grant CHE-1531468. PG would like to acknowledge funding from Innovation Fund Denmark for the AEngine Grand Solutions project.
Disclosure statement
No potential conflict of interest was reported by the author(s).