
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Understanding the interaction of the carboxylic group with TiO2 is crucial for photocatalytic degradation of pollutants, because aromatic molecules containing carboxylic acid groups are among the most common micropollutants. This study investigated the interactions of the anatase TiO2 (101) surface with several aromatic carboxylic acids: benzoic, nicotinic, salicylic and anthranilic acid, using dispersion-corrected density functional theory (DFT) calculations. For all the molecules studied, we found higher stabilities of monodentate adsorbed configurations over bidentate. Dispersion was found to have a significant effect on adsorption energies. In particular, dispersion gave rise to a tilted monodentate adsorption configuration, where adsorption through interfacial covalent and hydrogen bonds was additionally stabilised by dispersion interactions of the aromatic ring with the surface. Comparative calculations using DFT with empirical dispersion correction and van der Waals-corrected functionals found the relative stabilities of adsorbed structures to be independent of the method of describing dispersion. Thermodynamic probabilities of different adsorption configurations were evaluated using the Boltzmann distribution, and the tilted dispersion-stabilised structures were predicted to be by far the most abundant. Finally, the optical absorption of TiO2-acid systems was modelled, and TiO2-aromatic acid complexes were found to have their optical absorption extended into the visible range.
GRAPHICAL ABSTRACT
1. Introduction
Water pollution is an everyday growing environmental concern [Citation1]. Conventional wastewater treatment techniques, such as filtration and microbial treatment, cannot completely remove water micropollutants (defined as pollutants with the abundance of few micrograms per litre of water or with ppb level of concentration) [Citation2]. Therefore, advanced techniques are required that can destroy pollutants or convert them into non-toxic form. Photocatalytic treatment has emerged as a promising technique for destruction of organic contaminants in water and air [Citation3,Citation4]. In particular, TiO2 based photocatalysts are widely used for water treatment due to their low cost, stability, non-toxicity and good performance [Citation5]. One of the most common emerging micro-pollutants is salicylic acid (ortho-hydroxybenzoic acid), which is typically detected in wastewaters with concentrations up to 212 µg/L [Citation6–8]. Although these concentrations are well below its toxicity level [Citation9], salicylic acid in wastewater was reported to retain its pharmacological activity [Citation10]. Salicylic acid in wastewaters originates from various sources: it is present in industrial wastewaters as a key precursor in manufacturing of pharmaceuticals including aspirin and a range of other drugs [Citation11], in hospital wastewaters as a metabolite of aspirin [Citation7], and in domestic wastewaters as one of the key ingredients of skincare products and cosmetics [Citation12]. Aromatic carboxylic acids are also among the key intermediates in photocatalytic degradation of other pollutants, e.g. benzoic acid, salicylic acid and anthranilic acid are among the degradation intermediates of the carbamazepine drug [Citation13]; benzoic and anthranilic acid are also among degradation products of dyes, such as rhodamine B and indigo carmine [Citation14,Citation15]. Many other persistent micropollutants in water also contain the carboxylic group in their molecular structure, e.g. drugs such as clofibric acid [Citation16], ibuprofen [Citation17], diclofenac [Citation18], and herbicides such as mecoprop [Citation16]. Because of the prominence of carboxylic acids among micropollutants and the pressing need to develop photocatalytic methods for destroying these pollutants, it is important to understand the nature of binding of salicylic acid and other carboxylic acid molecules containing additional functional groups with the TiO2 photocatalyst surface. In particular, strong adsorption of micropollutant molecules on the photocatalyst surface facilitates their rapid photocatalytic degradation [Citation19]; at the same time, accumulation of degradation products or intermediates at the photocatalyst surface can lead to photocatalyst poisoning and may eventually make it non-functional [Citation20]. Therefore, understanding the nature and strength of binding of key pollutants, intermediates and products of degradation will help improve the efficiency of photocatalysis.
Moreover, besides removal of carboxylic acid pollutants, TiO2-carboxylic acid binding plays a role in photosensitisation of TiO2 for dye-sensitised solar cells (DSSC) [Citation21] and for photocatalysis [Citation22], where organic and metal–organic sensitiser molecules are used which typically contain a carboxylic acid anchoring group. For example, the same salicylic acid was used as a surface modifier on titania photocatalysts to enable visible light activity of the modified TiO2 photocatalysts to achieve visible light-driven oxidation of organics [Citation23] and degradation of pollutants [Citation24,Citation25]; this effect was attributed both to visible light absorption by the TiO2-salicylic acid composites [Citation23–25] and to increased binding of aromatic pollutants to salicylic acid-functionalised TiO2 [Citation24]. Thus, understanding the strength and nature of TiO2-carboxylic group interactions is essential for improving the efficiency of photocatalysts and solar cells.
Because of these important applications of TiO2-carboxylic acid systems, adsorption of carboxylic acids, such as formic, acetic and benzoic acid and dyes used in DSSCs, on different TiO2 polymorphs has been extensively studied experimentally and theoretically [Citation26–37]. However, despite this being a longstanding research topic, many questions remain, such as the exact nature of binding of carboxylic acids on the most stable anatase (101) surface [Citation36,Citation37]. Besides this, much less is known about adsorption of carboxylic acids containing additional functional groups, such as salicylic acid (containing a hydroxyl group) and anthranilic acid (containing an amine group), and aromatic rings besides benzene and perylene which are used in DSSC dyes. Moreover, since many of the theoretical studies of carboxylic acid adsorption were done in the early 2000s, they typically did not include the contribution of dispersion interactions. However, dispersion can have significant quantitative and qualitative effects, especially in the adsorption of aromatic molecules [Citation38–40]. Therefore, this paper aims to investigate the interactions of the thermodynamically most stable (101) surface of the anatase form of TiO2 with aromatic carboxylic acid pollutants containing additional functional groups (Figure ), such as salicylic acid and anthranilic acid, and an aromatic acid containing a heteroatom (nicotinic acid, which contains a pyridine ring and is an example of an N-heterocycle pollutant [Citation41]). We compare them with the well-studied adsorption of formic acid and benzoic acid on anatase (101), paying particular attention to the effect of the aromatic ring and the side groups in the aromatic ring. We investigate the effect of van der Waals interactions on the stabilities of the calculated structures, and compare the results obtained using different theoretical models of dispersion. We then evaluate the relative abundances of different adsorbed configurations of these molecules using the Boltzmann distribution, based on their adsorption energies. Finally, we simulate the light absorption of TiO2-carboxylic acid systems to investigate visible-light activity of these systems.
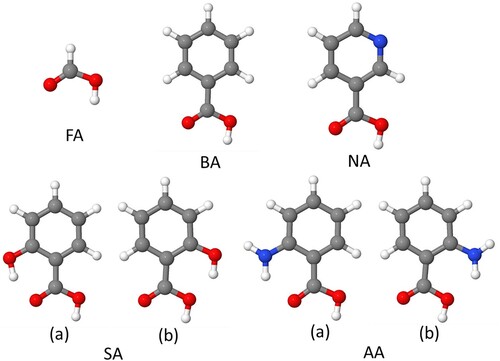
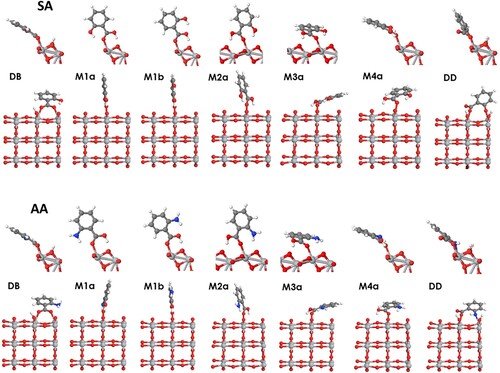
Figure 1. Structures of carboxylic acids modelled in this study: formic acid (FA), benzoic acid (BA), nicotinic acid (NA), salicylic acid (SA), and anthranilic acid (NA). Two configurations, (a) and (b), are shown for SA and AA. In this and the following figures, dark grey spheres show C atoms, red – O, blue – N, white – H, light grey – Ti atoms.
2. Method
Density functional theory (DFT) calculations were performed using CP2K software [Citation42–44]. Perdew–Burke–Ernzerhof (PBE) [Citation45] functional was used, with and without Grimme’s D3 dispersion correction [Citation46], in combination with double-ζ valence polarised (DZVP) basis sets [Citation47] and Goedecker–Teter–Hutter (GTH) pseudopotentials [Citation48]. Periodic slabs of TiO2 anatase with the (101) surface orientation and with the thickness of three TiO2 repeat layers were used. 1 × 3 extension of the surface unit cell was used (18 Ti and 36 O atoms), to accommodate the adsorbate molecules in the upright position. Carboxylic acids (formic acid, benzoic acid, nicotinic acid, salicylic acid, and anthranilic acid) were placed on the anatase (101) slabs in various orientations using tetr software [Citation49]. Multiple starting structures were considered for all configurations. To check the effect of the cell size on the adsorption structures and energies, stable adsorption configurations of formic and salicylic acid were additionally calculated in 2 × 3 and 1 × 6 extended surface unit cells (36 Ti and 72 O atoms), using PBE-D3. Geometry optimisations were carried out with all atoms free to move. Optimisation was carried out using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm, with the convergence criteria of 3 × 10−3 Bohr for the maximum geometry change, 4.5 × 10−4 Ha Bohr−1 for the maximum force, 1.5 × 10−3 Bohr for the root mean square geometry change, and 3 × 10−4 Ha Bohr−1 for the root mean square force.
Adsorption energies Eads were calculated according to the equation:
where E(slab) is the energy of the TiO2 anatase (101) slab, E(mol) is the energy of adsorbate molecule and E(slab + mol) is the energy of the slab-adsorbate system. Vibrational frequencies were calculated for the adsorption configurations of formic and salicylic acid; the frequencies for all structures were positive, confirming that these structures are energy minima.
Additional calculations of selected adsorption configurations of formic acid and salicylic acid were carried out using several different methods of describing dispersion interactions: pure DFT functionals with Grimme’s D3 [Citation46] dispersion correction (PBE-D3 [Citation45], revPBE-D3 [Citation50], B97-D3 [Citation51]), and non-local van der Waals-corrected functionals (vdW-DF [Citation52], vdW-DF2 [Citation53], optPBE [Citation54], optB88 [Citation54], C09x-vdW [Citation55], rVV10 [Citation56,Citation57]). For reference, exchange and correlation functionals used in these functionals, and dispersion corrections (where applicable), are summarised in Table S1 in the Supplementary Information.
Optical absorption of TiO2/adsorbate systems was calculated using linear-response time-dependent density functional theory, which was recently implemented in the CP2K software [Citation58]. Calculations for each system included 1000 excited states. Simulated absorption spectra were obtained by Gaussian broadening (σ = 0.2 eV) of the z-component of the transition dipole at each excitation energy. Molecular orbitals involved in the excitations, and projected densities of states, were also calculated within CP2K for the most stable configurations of each acid adsorbed on anatase (101).
3. Results and discussion
3.1. Choice of adsorption configurations
Adsorption configurations for formic, benzoic, nicotinic, salicylic and anthranilic acid were chosen by considering past literature studies of carboxylic acid adsorption and taking into account additional binding that may be possible in aromatic carboxylic acids containing additional functional groups. A number of literature studies investigated the adsorption of formic acid on the anatase (101) surface: experimental spectroscopic studies indicated co-existence of monodentate and bidentate adsorption [Citation32,Citation37]; computational studies modelled bidentate dissociative adsorption and monodentate (molecular and dissociative) adsorption [Citation26,Citation34,Citation35,Citation37]. Monodentate adsorption was typically found to be the most stable in density functional theory calculations [Citation26,Citation34,Citation35], although a recent study using the SCAN functional found bidentate dissociative adsorption to be more stable [Citation37]. The same types of structures (dissociative bidentate and molecular monodentate) were modelled for benzoic acid, with molecular monodentate shown to be the most stable structure [Citation27,Citation31].
For salicylic acid on anatase, experimental studies carried out using Fourier Transform Infrared Spectroscopy (FTIR) showed evidence of Ti-carboxylate complexation; as a result, a number of dissociative adsorption configurations were hypothesised, which involved binding through both the carboxylate and hydroxyl or hydroxylate groups [Citation59–61]. These dissociated chemisorbed structures were interpreted either as two co-existing adsorbed species [Citation61], or as initial non-dissociative adsorption followed by the final, stable dissociative adsorption, which was proposed to account for the sluggish adsorption of this molecule on anatase [Citation60]. Only limited theoretical evidence exists for these structures: one theoretical study considered three possible dissociated structures on a small anatase cluster, where a doubly dissociated structure bonded through both the carboxylate and the hydroxylate groups was favoured [Citation62].
Thus, a greater variety of adsorption configurations was proposed in the literature for salicylic acid than for the simpler acids, such as formic and benzoic acid. Similarly, complex adsorption can be expected for other carboxylic acids containing multiple functional groups, e.g. anthranilic acid (containing a carboxylic group and an amine group in the ortho-position); however, we are not aware of experimental or theoretical studies of adsorption configurations of anthranilic acid and similar ortho-substituted carboxylic acids on TiO2. Therefore, we modelled the adsorption of these molecules to get insight into the adsorption of aromatic carboxylic acids containing additional functional groups.
Based on the literature studies of formic, benzoic and salicylic acid and on our analysis of their possible bonding to the anatase (101) surface, the following adsorption configurations were modelled (optimised structures shown in Figure for formic acid (FA), Figure for benzoic acid (BA) and nicotinic acid (NA), and Figure for salicylic acid (SA) and anthranilic acid (AA)):
DB – Dissociative bridging-bidentate adsorption via the carboxylic group, with two carboxylate oxygens forming two covalent bonds to a pair of surface Ti atoms, and the hydrogen atom dissociated from the carboxylic group adsorbed nearby;
M1 – monodentate molecular adsorption via the carboxylic group, with the carboxylic oxygen forming a covalent bond to the surface titanium atom, and the hydrogen atom of the carboxylic group forming a hydrogen bond to the nearby surface oxygen;
M2 – monodentate molecular adsorption via the carboxylic group, with the carboxylic oxygen forming a covalent bond to the surface titanium atom, and the hydrogen atom of the carboxylic group forming a hydrogen bond to the surface oxygen in the neighbouring surface unit cell (rotated by ∼150° with respect to structure M1; note that two symmetrically equivalent M2 structures are possible, as shown in Figure );
M3 – monodentate molecular adsorption, similar to M2, with the plane of the aromatic ring tilted towards the surface rather than upright;
M4 – monodentate molecular adsorption in the same orientation as the dissociated structure DB (rotated by 90° with respect to structure M1), with the carboxylic oxygen forming a covalent bond to the surface titanium atom, and the hydrogen atom of the carboxylic group forming a hydrogen bond to the nearby oxygen that bridges two surface Ti atoms;
DD – bidentate adsorption of a doubly dissociated structure, adsorbed simultaneously via the carboxylate group and the deprotonated hydroxyl or amine group, based on the FTIR studies in Ref [Citation61]. (this structure is possible only for salicylic and anthranilic acid).
Figure 2. Adsorption of formic acid on anatase (101). Here and in the following figures, structures optimised using PBE-D3 are presented. Two possible symmetrically equivalent orientations of configuration M2 are shown.
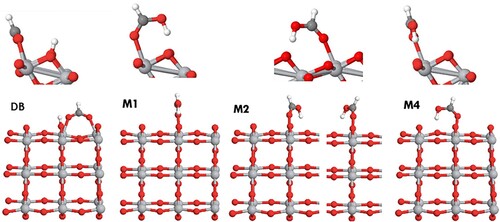
Figure 3. Adsorption of benzoic acid (BA, top row) and nicotinic acid (NA, bottom row) on anatase (101). Note that two symmetrically equivalent orientations of configurations M2 and M3 are possible (only one orientation was calculated for each).
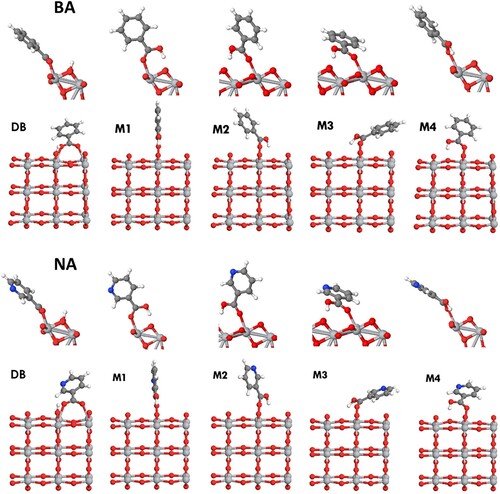
Figure 4. Adsorption of salicylic acid (SA, top row) and anthranilic acid (AA, bottom row) on anatase (101).
3.2. Stabilities of adsorption configurations
Adsorption of FA, BA, NA, SA and AA in these configurations on anatase (101) was calculated using PBE-D3 and PBE methods. The adsorption energies for structures optimised using PBE-D3 and using PBE are presented in Table and Figure , and the surface-adsorbate bond distances for these structures are summarised in Table . The adsorption energies for PBE-D3 optimised structures in the larger (2 × 3) and (1 × 6) extended surface unit cells are presented in Table S2. Primarily, PBE-D3 results will be discussed below, and key differences in PBE results will be highlighted.
Figure 5. Adsorption energies of carboxylic acids calculated with PBE-D3 (left) and PBE (right).
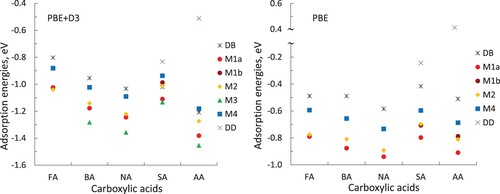
Table 1. Adsorption energies of formic acid, benzoic acid, nicotinic acid, salicylic acid and anthranilic acid on anatase (101), calculated using PBE-D3 for PBE-D3 optimised structures (energies calculated using PBE for PBE-optimised structures are given in brackets).
Table 2. Surface-adsorbate Ti-O bond and O … H hydrogen bond distances for formic acid, benzoic acid, nicotinic acid, salicylic acid and anthranilic acid adsorbed on anatase (101), optimised using PBE-D3 and PBE (PBE values are given in brackets).
3.2.1. Comparing carboxylic acids’ strengths of adsorption
Our results show that all of the investigated structures are strongly adsorbed, with large negative adsorption energies of the order of −1 eV. The adsorption energies are slightly (by 0.01-0.15 eV) more negative in the (2 × 3) and (1 × 6) extended surface unit cells (Table S2); however, the trends in adsorption stabilities are not affected by the cell size. We find that aromatic acid molecules (benzoic, nicotinic, salicylic and anthranilic acid) are more strongly adsorbed than formic acid. For example, for the dissociative bridging-bidentate structure DB, the strength of adsorption increases in the sequence FA < BA < SA ≤ NA < AA. A similar trend can be seen in the monodentate structures M1-M4, where the strength of adsorption increases as FA < SA < BA < NA < AA. This sequence of adsorption energies is consistent with the surface-adsorbate Ti-O distances (Table ), which are longer for adsorbed FA than for aromatic acids (except some SA configurations) by up to 0.10 Å. This trend in the strengths of adsorption can be attributed to the electron-withdrawing nature of the phenyl group, which strengthens the partly ionic bonding between carboxylic oxygens and surface Ti4+ ions and thus makes the adsorption of aromatic acids stronger. Additionally, the higher electronegativity of O and N than C makes the aromatic groups of NA, SA and AA more electron-withdrawing than BA, thus accounting for the stronger adsorption of NA, SA and AA. In particular, the adsorption configurations of nicotinic acid are very similar to benzoic acid, but nicotinic acid is systematically 0.06–0.08 eV more strongly adsorbed. This trend suggests that changes to the aromatic ring not next to the anchoring group do not have a qualitative effect on adsorption configuration, but they may affect the strength of adsorption.
The presence of hydroxyl or amine groups in the aromatic rings of salicylic and anthranilic acid has a greater effect on the stabilities of adsorbed structures. A larger variety of adsorption configurations is possible for SA and AA than for benzoic acid, because the SA and AA molecules have two stable isomers, where the hydroxyl group in SA and the amine group in AA can form intramolecular hydrogen bonds either with the carbonyl oxygen or with the hydroxyl oxygen of the carboxylic group (Figure ). This intramolecular hydrogen bonding affects the stabilities of the isomers: for isolated molecules, the isomers ‘a’ with hydrogen bonds to the carbonyl oxygen are more stable by 0.26 eV (salicylic acid) and 0.17 eV (anthranilic acid) than the isomers ‘b’. Consequently, each of the monodentate adsorbed configurations of salicylic and anthranilic acids can have two isomers, which differ by the position of the intramolecular hydrogen bond (by comparison, dissociated bidentate structures have only a single isomer, because the carboxylate oxygens are equivalent to each other). Therefore we modelled two isomers for structure M1 for salicylic and anthranilic acid, labelled as M1a and M1b. The structures M1a, involving hydrogen bonding between the hydroxyl or amine group and the carbonyl oxygen, were 0.12-0.19 eV more stable than the equivalent structures M1b, consistent with the results for isolated molecules. Therefore, for all other monodentate configurations (M2, M3 and M4), only the structures involving intramolecular hydrogen bonding to the carbonyl oxygen (similar to M1a) were considered for SA and AA.
Salicylic acid has unexpectedly rather weak monodentate adsorption compared to the other aromatic acids, 0.1-0.3 eV weaker than the corresponding adsorbed structures of BA, NA and AA. This can be attributed to the hydroxyl group forming a strong hydrogen bond (1.73–1.95 Å) with the carboxyl oxygen and therefore weakening the surface-adsorbate Ti-O bond. It can be illustrated by the bond distances shown in Table : the Ti-O distances for adsorbed SA are up to 0.08 Å longer than for adsorbed BA and NA, and up to 0.14 Å longer than for adsorbed AA. In contrast, the hydrogen bond between the amine hydrogen and the carboxylic oxygen in anthranilic acid are rather weak (1.98–2.08 Å) and does not significantly affect surface-adsorbate bonds; additionally, in structures M2 and M3 the amine group is able to form additional hydrogen bonds (2.05 Å in M2; 2.42 and 2.36 Å in M3) with the nearby surface oxygens, which contribute to the strong adsorption of this molecule.
3.2.2. Comparing stabilities of adsorption configurations.
Comparison of the energies of different adsorption configurations in Table and Figure shows that, for all adsorbates considered in this study, monodentate molecular adsorption is stronger than dissociative bidentate adsorption by 0.1–0.3 eV. Salicylic acid is the only adsorbate where the monodentate configurations are close in stability (within 0.11 eV) to the dissociated bidentate configurations, probably because the hydrogen bonding between the hydroxyl and carboxyl groups stabilises the dissociated acid form, unlike the other acids where the monodentate configurations are clearly more stable. This greater stability of the monodentate configurations is in agreement with the previous computational studies of formic acid [Citation26,Citation34,Citation35] and other carboxylic acids, e.g. benzoic acid [Citation27,Citation31], acetic acid [Citation30], perylenecarboxylic acid [Citation33] and cyanoacrylic acid [Citation31] on the anatase (101) surface modelled using GGA and hybrid functionals, which similarly found molecular monodentate adsorption to be stronger than bidentate dissociative adsorption (however, the opposite trend for formic acid adsorption configurations was reported in a recent study using the SCAN functional [Citation37]). In contrast, experimental infrared spectroscopy studies of formic acid on anatase reported dissociative adsorption of the deprotonated formate species [Citation32]; however, this contradiction was recently explained as monodentate adsorption with the carboxylic hydrogen being able to shuttle between the carboxylic group and the surface oxygen [Citation36], while the spectroscopic observation of the deprotonated bidentate structure was attributed to the presence of subsurface oxygen vacancies, which favour deprotonation and formation of bidentate structures [Citation32,Citation37]. A recent experimental study [Citation37] reported that formic acid adsorbed on anatase (101) initially in the monodentate form at low temperatures (80 K), and then converted to bidentate upon annealing, suggesting higher stability of the bidentate form. Our results are more consistent with the low-temperature results in Ref. [Citation37] where the monodentate form dominates, possibly indicating entropic stabilisation of the bidentate form at higher temperatures.
Our results show that the monodentate structures M1 and M2 have very similar stabilities for all acids. In formic acid, both of these structures are equally stable (within 0.02 eV), while the aromatic molecules have a slight preference (up to 0.11 eV) for the structure M1, where the carboxylic hydrogen atom forms a hydrogen bond to the surface oxygen atom that is next to the titanium atom which forms the Ti-O interfacial bond.
M4 is the least strongly adsorbed monodentate structure for all acid molecules: it is less strongly adsorbed than M1 and M2 by up to 0.20 eV and is less strongly adsorbed than M3 (which is discussed in more detail below) by up to 0.27 eV, and is comparable in stability to the bidentate structure DB. For all adsorbates, M4 structures have consistently longer surface-adsorbate bonds than M1-M3: up to 0.2 Å longer Ti-O bonds, and as much as 0.47 Å longer O … H bonds, which confirm that surface-adsorbate bonding in M4 is quite strained and is less favourable than in the other monodentate structures. The structures M4 and DB are in similar positions on the anatase (101) surface (Figures 2–4) and differ only in the nature of binding, therefore these two structures offer the most likely path for conversion between monodentate and bidentate adsorption. In particular, in salicylic and anthranilic acid M4 is slightly less stable than DB (by 0.03-0.08 eV), therefore conversion from M4 to DB would be favourable.
To evaluate the effect of dispersion on the strength of adsorption, we compared the adsorption energies of all structures obtained in PBE-D3 and PBE calculations (Figure ). As expected, the calculations with the dispersion correction predict stronger adsorption. The difference between PBE-D3 and PBE adsorption energies increases with the size of the molecule: PBE-D3 values are more favourable by 0.21−0.31 eV for formic acid, by 0.30−0.46 eV for benzoic and nicotinic acid, and by 0.28−0.70 eV for salicylic and anthranilic acid. These results show that dispersion interaction makes a non-negligible contribution to the strength of binding of these chemisorbed structures. Ti-O and O … H bond energies are very similar in PBE and PBE-D3 calculations (PBE values usually only up to 0.04 Å longer, see Table ), therefore this change in energies is not due to an increase in covalent bond strength but due to non-bonded interactions between the surface and the adsorbates. However, the qualitative trends in both the pure DFT and dispersion-corrected DFT results are, for the most part, very similar: both methods favour monodentate structures over bidentate structures for all considered molecules, with similar ordering or structures, although the energy differences between structures are smaller in PBE-D3 than in PBE calculations. As seen in Figure , according to PBE calculations, molecular monodentate configuration M1 is the most stable adsorption configuration for these carboxylic acids; however, in a key difference, this configuration is the second most stable in PBE-D3 calculations.
A new and very stable adsorption configuration M3 was obtained in PBE-D3 calculations of aromatic acids, which was not obtained in PBE calculations and was not found in previous studies of carboxylic acids on TiO2. The configuration labelled M3 (Figures and ) is based on the configuration M2, so that the chemical bonds formed between the adsorbates and the surface are the same in both configurations: an interfacial Ti-O bond and an O … H hydrogen bond; however, the structures M2 are upright (similar to the structures calculated in the previous studies of BA adsorption [Citation27,Citation31]), while the structures M3 have the aromatic ring tilted towards the surface. The configurations M3 are more strongly bound than M2 by 0.1–0.2 eV for all aromatic acids, and are the most stable of all adsorption configurations considered in this work. Since no new covalent or hydrogen bonds are formed, this increased strength of adsorption can be fully attributed to the additional dispersion interaction between the aromatic ring of the adsorbate and the surface, when the aromatic ring is in close proximity to the surface. Thus, these structures could not be obtained in pure DFT calculations which do not properly account for dispersion interactions. The structures M2 can be seen as local energy minima, which provide strong interfacial Ti-O and O … H bonds (as shown in Table ), while structures M3 have slightly longer interfacial Ti-O and O … H bonds (up to 0.04 Å longer Ti-O and up to 0.10 Å longer O … H than in M2), and are stabilised by the additional dispersion interactions of the aromatic ring with TiO2. Therefore, dispersion certainly plays an important role in adsorption of aromatic carboxylic acids: besides making a contribution to the overall adsorption energy (as seen by comparing PBE and PBE-D3 adsorption energies), it makes a qualitative difference and gives rise to the new very stable tilted configuration M3.
In contrast, structure M3 is not possible for formic acid, which lacks an aromatic ring and therefore cannot be stabilised by tilting towards the surface. An equivalent tilted configuration was not obtained for M1, probably because it would have involved unfavourable strain on the interfacial bonds. To the best of our knowledge, there are no experimental data as yet that could confirm which of the structures, either the tilted configuration M3 or the upright configurations M1/M2, are formed by aromatic acids on anatase (101), but it may be possible to differentiate them using Near Edge X-ray Absorption Fine Structure (NEXAFS) spectroscopy [Citation63,Citation64] or infrared reflection absorption spectroscopy (IRRAS) [Citation32]. We hypothesise that the configuration M3 is likely to form at low coverages (the situation most relevant to micropollutants in water, which are present in low concentrations), because it offers the largest binding energy per molecule, while M1 or M2 are more likely to form at higher coverages, because they allow a larger number of adsorbates per unit area.
Finally, we modelled the doubly dissociated configuration DD which is adsorbed via two functional groups: carboxyl and hydroxyl (for salicylic acid) or carboxyl and amine (for anthranilic acid), as proposed in the experimental analysis of salicylic acid adsorption [Citation59–61]. In this configuration, both the carboxylic and the hydroxyl or amine groups were dissociated; although one of the carboxylic oxygens maintained a hydrogen bond with a proton adsorbed nearby on the surface. However, these structures were the least stable ones both for salicylic acid and especially for anthranilic acid, where DD was 0.67 eV less stable than the next least stable structure M4. These results suggest that dissociation of the hydroxyl and especially of the amine side groups and adsorption through these groups is not favourable. However, participation of the hydroxyl side group in adsorption may still be possible for TiO2 surfaces involving vacancies, steps or edges.
Overall, our calculations show that the adsorption configurations of salicylic and anthranilic acid are very similar to benzoic and nicotinic acid, but their strength of adsorption is affected by the side groups. In particular, the calculated stabilities of adsorbed salicylic acid structures can help explain the results of experimental spectroscopic and reaction kinetics studies of salicylic acid on anatase, which found evidence of several structures with multiple coordination of salicylic acid to TiO2 [Citation60,Citation61]. Conversion from an initial adsorbed structure with fast adsorption kinetics to a more stable structure with slower adsorption kinetics, which was proposed in Ref. [Citation60], can be interpreted as conversion from the monodentate structure M4 to the structurally similar but slightly more stable structure DB. The presence of two different adsorbed structures of salicylic acid on TiO2, which was proposed in Ref. [Citation61], can be interpreted as co-existence of one or several of the monodentate structures M1-M3 with the bidentate structure DB.
It should be noted that molecules such as carboxylic acids in aqueous solution maintain an equilibrium between their dissociated and non-dissociated forms, dependent on the solution pH, which can affect their adsorbed structures. For example, experimental studies of cysteine and citric acid adsorption on TiO2 demonstrated the adsorption of these molecules in different protonation states, dependent on the solution pH [Citation65,Citation66]. In our study we considered adsorption of protonated carboxylic acid molecules, the dominant form at low pH, while at medium and high pH the dissociated form would be present in larger amount and the dissociated adsorption configurations are likely to be favoured. The protonation state of TiO2 surfaces was also reported to depend on the solution pH [Citation67,Citation68]. Therefore, for a comprehensive description of adsorption equilibria, it would be worthwhile to investigate the binding of these adsorbates on the protonated and hydroxylated TiO2 surfaces and the kinetics of exchange of adsorbed surface hydroxyls for adsorbed acids; however, this is beyond the scope of this work.
3.3. Stabilities of adsorption configurations: comparison of density functionals and theoretical models for van der Waals interactions
The results presented in the previous section clearly show that the dispersion correction affects the stabilities of adsorbed structures and gives rise to a new adsorption configuration M3. Previous computational studies of the model molecule formic acid on anatase (101), carried out using pure or hybrid DFT functionals with or without dispersion correction, typically favoured monodentate adsorption configurations; however, slightly different energy differences between monodentate and bidentate configurations were reported, ranging between 0.10-0.24 eV [Citation26,Citation34,Citation35]; the opposite trend with preference for bidentate adsorption was also recently reported [Citation37]. Thus, it is worthwhile to investigate how the stabilities of different adsorption configurations depend on the choice of the DFT functional and on the method of describing dispersion interactions. Therefore, we compared adsorption energies of selected configurations using several dispersion-corrected DFT methods (listed in Section 2 and summarised in Table S1): pure DFT functionals with Grimme’s D3 dispersion correction (PBE-D3, revPBE-D3, B97-D3), and non-local van der Waals-corrected functionals (optPBE, optB88, C09x-vdW, vdW-DF, vdW-DF2, rVV10). We modelled formic acid as the classic well-studied case, and salicylic acid as the ‘difficult’ case because of the presence of the hydroxyl group in the aromatic ring. Based on the stabilities of the structures identified in the previous section, three adsorption configurations were compared for each molecule: the dissociated bidentate configuration DB, the monodentate molecular configuration M1 (M1a for salicylic acid), and the monodentate tilted configuration M3 for salicylic acid (and its non-tilted analogue M2 for formic acid).
The results are presented in Figure . There are some quantitative differences in the values of adsorption energies, e.g. revPBE-D3 consistently gives the least negative adsorption energies, while C09x-vdW gives the most negative energies. However, all investigated density functionals and all models for van der Waals interactions find monodentate adsorption to be more stable than bidentate, although the difference is larger in the calculations that use Grimme’s dispersion correction (0.11–0.53 eV difference) than in the calculations that use non-local functionals (0.01–0.18 eV difference). All considered methods show very slight preference for the structure M2 over M1 for formic acid (by 0.01–0.04 eV, except C09x-vdW, where M1 is only 0.01 eV more stable). All methods except revPBE-D3 show a slight preference (0.02–0.11 eV) for the tilted structure M3a over M1a for salicylic acid. Thus, all investigated density functionals and models for van der Waals interactions give a very similar picture of the relative stabilities of these molecules’ adsorption structures. Therefore, the trends in stabilities discussed in the previous section are confirmed as reliable within the accuracy of DFT: monodentate adsorption preferred over bidentate, and the tilted monodentate configuration preferred over upright.
Figure 6. Interaction energies of three conformations of formic acid (upper panel) and salicylic acid (lower panel) adsorbed on anatase (101), calculated using different dispersion-corrected DFT functionals.
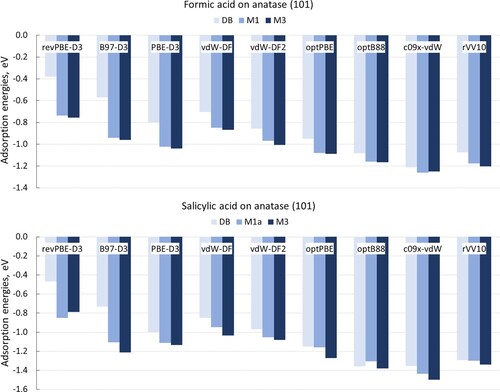
These data also show that all density functionals compared in this study produce consistent results, and therefore the trends in stabilities of adsorbates can be considered robust and not dependent on the functional or on the method for describing van der Waals interactions. Therefore, according to our results, it is not possible to recommend one or few best functionals for modelling acids adsorption on TiO2, but instead, any van der Waals-corrected density functional scheme can be reliably used for modelling carboxylic acid adsorbates on TiO2.
3.4. Relative abundances of adsorbed configurations
The calculations described in the previous section identified the most stable configurations for each of the acid adsorbates on anatase (101). However, the less stable configurations were only few tenths of eV above the most stable configurations. In some cases, such as formic and salicylic acid, the differences between the most stable and second most stable configurations were less than 0.1 eV. In such situations, it can be expected that several adsorption configurations can co-exist. Therefore, we quantified relative abundances of different adsorption configurations for each acid on TiO2, similar to a recent study of thermodynamic distribution of defects in uranium oxide [Citation69], using the Boltzmann distribution formula:
Here Pi is the probability of configuration i, and
is defined as
where
is the degeneracy of configuration i, Ei is the energy of configuration i relative to the energy of the most stable configuration, R is the universal gas constant and T is the temperature. The probabilities were evaluated at the temperature of 298 K, using the energies calculated at the PBE-D3 level (Table ). Configurations DB and M1 were considered to have the degeneracy of 1, while configurations M2, M3, M4 and DD were considered to have the degeneracy of 2, because each of them has two symmetrically equivalent orientations (as shown in Figure for configuration M2 for formic acid).
The resulting probabilities of configurations of formic, benzoic, nicotinic, salicylic and anthranilic acid on anatase (101) are shown in Figure . The figure shows that, while there is a dominant adsorption configuration for each acid, other adsorption configurations are likely to be present in small amounts. For all acids, monodentate adsorption configurations dominate, while bidentate configurations are present in negligible amounts. For formic acid, two monodentate configurations are predicted to exist in appreciable amounts: the monodentate configuration M2 is present with the abundance of ∼79%, while a similar configuration M1, which is only 0.02 eV higher in energy, is predicted to have the abundance of ∼21%. These two dominant configurations have very similar interfacial bonding (both have an interfacial Ti-O bond and an O … H bond to a two-coordinated surface oxygen) and are likely to be indistinguishable spectroscopically.
Figure 7. Thermodynamic probabilities of different adsorption configurations of formic, benzoic, nicotinic, salicylic and anthranilic acid on anatase (101) at 298 K.
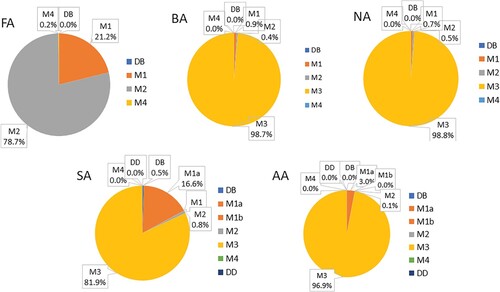
For all aromatic carboxylic acids considered in this work, the dominant adsorption configuration is the monodentate tilted configuration M3, which is present with a 97–99% abundance for BA, NA and AA, and with a 82% abundance for SA. Salicylic acid, which has small differences between the energies of different adsorption configurations (Table ), shows the richest variety of adsorption configurations: besides the tilted configuration M3, the upright configuration M1a is predicted to occur with a ∼17% probability, and there are small (<1%) probabilities of occurrence of another upright monodentate configuration, M2, and the bidentate dissociated configuration DB.
3.5. Light absorption of TiO2/adsorbed acid systems
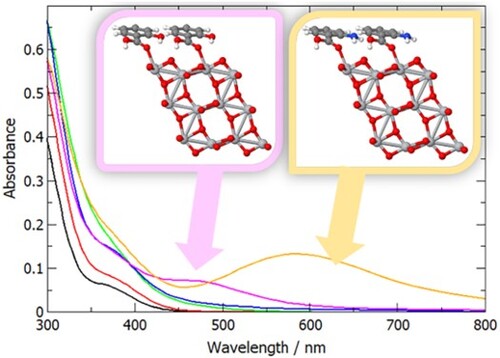
One of the interesting properties of TiO2/salicylic acid composite systems reported in the literature is the broadening of the optical absorption compared to pure TiO2 [Citation23–25]. While TiO2 absorbs only in the ultraviolet range, TiO2 particles functionalised by adsorbing salicylic acid were reported to be yellow in colour and absorb light up to 500 nm [Citation23,Citation24]; similarly, the absorption edge of TiO2 nanotubes was found to be red-shifted from 3.33 eV to 3.18 eV for SA-modified TiO2 nanotubes [Citation25]. To investigate the origin of this red shift and to verify whether it occurs with other carboxylic acids, we modelled the optical absorption of pure anatase (101) slabs, and the same slabs with formic, benzoic, nicotinic, salicylic and anthranilic acid adsorbed in their most stable configurations (M2 for formic acid, and M3 for aromatic acids).
The calculated optical absorption spectra are shown in Figure . All anatase/adsorbate systems show enhanced optical absorption in the long-wavelength region. Even the smallest adsorbate, formic acid, leads to slightly higher absorption intensity of the anatase/formic acid system, although the position of the absorption edge is unchanged. Presence of aromatic acid adsorbates results in the range of absorption extended towards longer wavelengths. This broadening of optical absorption is slightly greater for the aromatic group containing a heteroatom (nicotinic acid) than for benzoic acid, and it is particularly pronounced for the aromatic groups containing additional functional groups in the aromatic ring – salicylic acid and especially anthranilic acid adsorbates, where absorption across the whole visible range is predicted.
Figure 8. Calculated optical absorption spectra of anatase (101) with adsorbed formic, benzoic, nicotinic, salicylic and anthranilic acid, compared to anatase (101) without adsorbates.
These changes in the optical absorption can be interpreted with the help of projected densities of states (PDOS) of TiO2/adsorbate systems (Figure S1). The PDOS plots show the conduction band dominated by Ti and the valence band dominated by O of TiO2, with adsorbate states at or above the valence band maximum of all the TiO2/adsorbate systems. In the TiO2/formic acid system, the adsorbate’s highest occupied state is present just below the top of the valence band, which explains why absorption intensity is slightly enhanced but the position of the absorption edge is unchanged. The adsorbed benzoic acid has an occupied state at the top of the valence band, and adsorbed nicotinic acid – an occupied state just above the valence band; therefore, the broadening of the absorption can be attributed to new transitions from these highest occupied adsorbate states to the unoccupied Ti states. The adsorbed salicylic and anthranilic acid have their highest occupied states in the band gap, 1–1.5 eV above the valence band edge. Transitions from these highest occupied adsorbate states to the manifold of TiO2 unoccupied states can explain the broadened adsorption in the visible range for TiO2/salicylic acid and TiO2/anthranilic acid systems.
The nature of photoexcitation transitions can be further clarified by considering the orbitals involved in excitations. Table S3 summarises the key transitions in the TiO2/adsorbed carboxylic acids systems: the lowest-energy transitions responsible for the absorption edge, the lowest-energy direct TiO2→TiO2 excitations (if different from the lowest-energy transitions), and the transitions responsible for the peaks appearing in the visible range (the peak at ∼590 nm for TiO2/anthranilic acid and the shoulder peak at ∼470 nm for TiO2/salicylic acid). The data in Table S3 show that the lowest-energy excitations in all the studied anatase/aromatic acid systems are charge-transfer excitations from the highest occupied orbitals localised on the adsorbate to the lowest unoccupied orbitals delocalised over Ti atoms of TiO2. This is different from pure anatase (101) and the anatase/formic acid system, where the lowest-energy excitations are direct TiO2→TiO2 excitations. Besides the lowest-energy excitation, several of the low-energy excitations in TiO2/aromatic acid systems are similarly charge-transfer excitations, with direct TiO2→TiO2 transitions appearing only at the 3rd excitation (TiO2/BA), 13th (TiO2/NA), 30th (TiO2/SA) or 48th (TiO2/AA) excitations. Thus, these low-energy charge-transfer excitations are responsible for the broadening of the optical absorption of the TiO2/aromatic acid systems. Moreover, TiO2/SA and especially TiO2/AA have charge-transfer excitations with high transition dipole moments at the energies either just above the lowest-energy direct excitation (for TiO2/SA) or below the lowest-energy direct excitation (for TiO2/AA); these excitations are responsible for the prominent peaks at ∼470 nm and ∼590 nm observed in the calculated spectra of TiO2/SA and TiO2/AA.
This evidence for charge-transfer photoexcitation from PDOS spectra and molecular orbitals involved in excitations supports the hypothesis put forward in the experimental studies of TiO2 functionalised with salicylic acid, which proposed that the enhanced light absorption in these complexes is caused by ligand-to-metal charge transfer [Citation23,Citation60].
Thus, all TiO2/adsorbed aromatic acid systems studied in this work have a broader absorption range than pure anatase, extending into the visible region of the solar spectrum, as a result of charge-transfer excitations from the adsorbate’s occupied states to the unoccupied states of TiO2. This behaviour is similar to enhanced light absorption in TiO2/dye systems used in dye-sensitised solar cells [Citation28]. These excitations are likely to have a beneficial effect on the photocatalytic activity of these materials: absorption in a broader wavelength range would improve the light harvesting ability and therefore enhance the photocatalytic efficiency of adsorbate-functionalised TiO2. Moreover, since these charge-transfer excitations produce photoexcited electrons in the TiO2 conduction band but no holes in the valence band (the holes are instead on the adsorbate), electron–hole recombination in TiO2 would be reduced, and this can be expected to result in longer lifetimes of photoelectrons, again leading to increased efficiency of photocatalytic reduction processes. This is consistent with the experimental studies showing enhanced photocatalytic activity of salicylic acid-modified TiO2 compared to pure TiO2 [Citation23–25].
4. Conclusions
Our computational study investigated a variety of possible adsorption configurations of carboxylic acids on anatase (101), focussing on the effect of the aromatic ring and substituents in the aromatic ring on the nature of adsorption and on the surface/adsorbate systems’ light absorption. The adsorbates were chosen as the smallest relevant examples of persistent organic pollutants that may be present in wastewaters. Our calculations showed that aromatic carboxylic acids, such as benzoic acid, nicotinic acid, salicylic acid and anthranilic acid, adsorb more strongly than formic acid, even though the nature of covalent and hydrogen bonding at the surface-adsorbate interface is the same.
We show that the preference for monodentate adsorption configurations over bidentate, which was previously reported for formic and benzoic acid, also holds for aromatic acids containing heteroatoms and functional groups. Comparison of the results obtained with and without dispersion correction shows that dispersion contributes to the strength of adsorption of all configurations. Moreover, we find that dispersion leads to the most stable monodentate configuration where the adsorbate’s aromatic ring is tilted towards the surface. This configuration was not found in previous computational studies of carboxylic acids adsorption on TiO2. Its high stability is due to dispersion interactions between the aromatic ring and the TiO2 surface. This configuration is particularly relevant for situations of low coverage by adsorbates, e.g. for photocatalytic removal of micropollutant adsorbates, which are present in wastewaters in small concentrations. We compared our results with available computational and experimental data; in particular, spectroscopic observations of several adsorption modes for salicylic acid on TiO2 may be explained by conversion between monodentate and bidentate configurations.
To verify the reliability of these calculated stabilities of adsorption configurations and to investigate possible dependence on the choice of the computational method, we compared adsorption energies of several representative adsorption configurations for formic acid and salicylic acid using a series of DFT functionals with Grimme's dispersion correction and non-local (van der Waals) functionals. The same trends in stabilities were obtained with all considered DFT methods: monodentate adsorption was more stable than bidentate, and tilted adsorption configurations were more stable than upright ones.
We evaluated the relative abundances (thermodynamic probabilities of formation) of these adsorption configurations for each acid and found that, for each adsorbate, the dominant monodentate adsorption configuration is expected to be present with the abundance of 79–99% at room temperature. Other configurations may be present in small amounts, e.g. salicylic acid is predicted to have several configurations co-existing, but these configurations are primarily also of the monodentate type. This confirms that monodentate adsorption dominates on the clean anatase (101) surface. Alternative configurations, such as bidentate, may be preferred on stepped and defect-containing surfaces, which were outside the scope of this work.
Finally, we showed that adsorption of aromatic acids (in particular, salicylic and anthranilic acid which contain additional functional groups in the aromatic ring) extends the optical absorption of anatase to the visible range, as a result of charge-transfer excitations from the adsorbate’s occupied states to the unoccupied states of TiO2. These results suggest that relatively simple aromatic carboxylic acid molecules, such as salicylic acid, can be used to enhance light harvesting by TiO2 nanoparticles, as a low-cost way to improve the efficiency of photocatalysis.
TMPH-2022-0516-File012.docx
Download MS Word (13.6 MB)Acknowledgements
The authors acknowledge the University of Sheffield high performance computing (HPC) services (Iceberg and ShARC HPC clusters). Authors would like to thank Prof. Siddharth Patwardhan and Prof. Julia Weinstein (University of Sheffield) for their comments and advice on this work. The authors thank Dr Joseph Flitcroft (University of Sheffield, now at the University of Manchester) for advice on calculating thermodynamic probabilities, and Dr Matt Watkins (University of Lincoln) for advice on optical absorption calculations.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- D.G. Joakim Larsson, Philos Trans R Soc B Biol Sci. 369 (1656), 20130571 (2014).
- D. Kanakaraju, B.D. Glass and M. Oelgemöller, J. Environ. Manag. 219, 189 (2018).
- S. Malato, P. Fernández-Ibáñez, M.I. Maldonado, et al., Catal. Today. 147 (1), 1 (2009).
- J.J. Rueda-Marquez, I. Levchuk, P.F. Ibañez, et al., J. Cleaner Prod. 258, 120694 (2020).
- K. Nakata and A. Fujishima, J. Photochem. Photobiol., C. 13 (3), 169 (2012).
- C. Miège, J.M. Choubert, L. Ribeiro, et al., Environ. Pollut. 157 (5), 1721 (2009). doi:10.1016/j.envpol.2008.11.045.
- C.I. Kosma, D.A. Lambropoulou and T.A. Albanis, Sci. Total Environ. 466-467, 421 (2014). doi:10.1016/j.scitotenv.2013.07.044
- P.D. Scott, M. Bartkow, S.J. Blockwell, et al., J. Environ. Qual. 43 (5), 1702 (2014). doi:10.2134/jeq2014.01.0012.
- R.K. Madan and J. Levitt, J. Am. Acad. Dermatol. 70 (4), 788 (2014).
- R. López-Serna, A. Jurado, E. Vázquez-Suñé, et al., Environ. Pollut. 174, 305 (2013). doi:10.1016/j.envpol.2012.11.022
- D. Camacho-Muñoz, J. Martín, J.L. Santos, et al., Chemosphere. 111, 70 (2014). doi:10.1016/j.chemosphere.2014.03.043.
- E. Mikami, T. Goto, T. Ohno, et al., J. Pharm. Biomed. Anal. 28 (2), 261 (2002). doi:10.1016/S0731-7085(01)00564-7.
- M. Nawaz, W. Miran, J. Jang, et al., Appl. Catal. B Environ. 203, 85 (2017). doi:10.1016/j.apcatb.2016.10.007.
- M. Vautier, C. Guillard and J.-M. Herrmann, J. Catal. 201 (1), 46 (2001). doi:10.1006/jcat.2001.3232.
- L. Liang, L. Cheng, Y. Zhang, et al., RSC Adv. 10 (48), 28509 (2020). doi:10.1039/D0RA03125A.
- H.-R. Buser, M.D. Müller and N. Theobald, Environ. Sci. Technol. 32 (1), 188 (1998).
- N. Lindqvist, T. Tuhkanen and L. Kronberg, Water Res. 39 (11), 2219 (2005). doi:10.1016/j.watres.2005.04.003.
- P. Schröder, B. Helmreich, B. Škrbić, et al., Environ. Sci. Pollut. Res. 23 (13), 12835 (2016).
- H. Dong, G. Zeng, L. Tang, et al., Water Res. 79, 128 (2015). doi:10.1016/j.watres.2015.04.038.
- E. Barraud, F. Bosc, D. Edwards, et al., J. Catal. 235 (2), 318 (2005). doi:10.1016/j.jcat.2005.08.017.
- A. Hagfeldt, G. Boschloo, L. Sun, et al., Chem. Rev. 110 (11), 6595 (2010).
- J.-F. Huang, Y. Lei, T. Luo, et al., ChemSusChem. 13 (22), 5863 (2020). doi:10.1002/cssc.202001646.
- X. Li, H. Xu, J.-L. Shi, et al., Appl. Catal., B. 244, 758 (2019).
- S.X. Li, F.Y. Zheng, W.L. Cai, et al., J. Hazard. Mater. 135 (1-3), 431 (2006). doi:10.1016/j.jhazment.2005.12.010.
- X. Wang, H. Zhao, X. Quan, et al., J. Hazard. Mater. 166 (1), 547 (2009). doi:10.1016/j.jhazmat.2008.11.066.
- A. Vittadini, A. Selloni, F.P. Rotzinger, et al., J. Phys. Chem. B. 104 (6), 1300 (2000).
- N. Martsinovich, D.R. Jones and A. Troisi, J. Phys. Chem. C. 114 (51), 22659 (2010).
- F. De Angelis, S. Fantacci, E. Mosconi, et al., J. Phys. Chem. C. 115 (17), 8825 (2011). doi:10.1021/jp111949a.
- A.G. Thomas and K.L. Syres, Chem. Soc. Rev. 41 (11), 4207 (2012). doi:10.1039/C2CS35057B.
- C. Anselmi, E. Mosconi, M. Pastore, et al., Phys. Chem. Chem. Phys. 14 (46), 15963 (2012).
- E. Mosconi, A. Selloni and F. De Angelis, J. Phys. Chem. C. 116 (9), 5932 (2012).
- M. Xu, H. Noei, M. Buchholz, et al., Catal. Today. 182 (1), 12 (2012).
- S. Ikäläinen and K. Laasonen, Phys. Chem. Chem. Phys. 15 (28), 11673 (2013).
- L. Kou, T. Frauenheim, A.L. Rosa, et al., J. Phys. Chem. C. 121 (32), 17417 (2017).
- Y. Li and Y. Gao, Langmuir. 34 (1), 546 (2018).
- G. Tabacchi, M. Fabbiani, L. Mino, et al., Angew. Chem., Int. Ed. 58 (36), 12431 (2019).
- Y. Wang, B. Wen, A. Dahal, et al., J. Phys. Chem. C. 124 (37), 20228 (2020).
- S.D. Chakarova-Käck, E. Schröder, B.I. Lundqvist, et al., Phys. Rev. Lett. 96 (14), 146107 (2006). doi:10.1103/PhysRevLett.96.146107.
- P. João, P. Ramalho, J.R.B. Gomes and F. Illas, RSC Adv. 3 (32), 13085 (2013). doi:10.1039/C3RA40713F.
- W. Heckel, T. Würger, S. Müller, et al., J. Phys. Chem. C. 121 (32), 17207 (2017).
- K.V. Padoley, S.N. Mudliar and R.A. Pandey, Bioresour. Technol. 99 (10), 4029 (2008). doi:10.1016/j.biortech.2007.01.047.
- CP2K. Open Source Molecular Dynamics. www.cp2k.org.
- J. VandeVondele, M. Krack, F. Mohamed, et al., Comput. Phys. Commun. 167 (2), 103 (2005).
- T.D. Kühne, M. Iannuzzi, M. Del Ben, et al., J. Chem. Phys. 152 (19), 194103 (2020). doi:10.1063/5.0007045.
- J.P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett. 77 (18), 3865 (1996). doi:10.1103/PhysRevLett.77.3865.
- S. Grimme, J. Antony, S. Ehrlich, et al., J. Chem. Phys. 132 (15), 154104 (2010). doi:10.1063/1.3382344.
- J. VandeVondele and J. Hutter, J. Chem. Phys. 127 (11), 114105 (2007). doi:10.1063/1.2770708.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B. 54 (3), 1703 (1996). doi:10.1103/PhysRevB.54.1703.
- L.N. Kantorovich, LEV00 & TETR: user-friendly packages for DFT codes VASP / SIESTA / QUICKSTEP / CRYSTAL / Quantum Espresso.
- Y. Zhang and W. Yang, Phys. Rev. Lett. 80 (4), 890 (1998). doi:10.1103/PhysRevLett.80.890.
- S. Grimme, J. Comput. Chem. 27 (15), 1787 (2006). doi:10.1002/jcc.20495.
- M. Dion, H. Rydberg, E. Schröder, et al., Phys. Rev. Lett. 92 (24), 246401 (2004). doi:10.1103/PhysRevLett.92.246401.
- K. Lee, É.D. Murray, L. Kong, et al., Phys. Rev. B. 82 (8), 081101 (2010). doi:10.1103/PhysRevB.82.081101.
- J. Klimeš, D.R. Bowler and A. Michaelides, J. Phys.: Condens. Matter. 22 (2), 022201 (2009). doi:10.1088/0953-8984/22/2/022201.
- V.R. Cooper, Phys. Rev. B. 81 (16), 161104 (2010). doi:10.1103/PhysRevB.81.161104.
- O.A. Vydrov and T. Van Voorhis, J. Chem. Phys. 133 (24), 244103 (2010). doi:10.1063/1.3521275.
- R. Sabatini, T. Gorni and S. de Gironcoli, Phys. Rev. B. 87 (4), 041108 (2013). doi:10.1103/PhysRevB.87.041108.
- A.-S. Hehn, B. Sertcan, F. Belleflamme, et al., J. Chem. Theory Comput. 18 (7), 4186 (2022). doi:10.1021/acs.jctc.2c00144.
- J. Moser, S. Punchihewa, P.P. Infelta, et al., Langmuir. 7 (12), 3012 (1991).
- A.E. Regazzoni, P. Mandelbaum, M. Matsuyoshi, et al., Langmuir. 14 (4), 868 (1998).
- A.D. Weisz, L.G. Rodenas, P.J. Morando, et al., Catal. Today. 76 (2-4), 103 (2002). doi:10.1016/s0920-5861(02)00210-9.
- C.I. Oprea, P. Panait and M.A. Gîrţu, Beilstein J. Nanotechnol. 5, 1016 (2014). doi:10.3762/bjnano.5.115.
- A.G. Thomas, M.J. Jackman, M. Wagstaffe, et al., Langmuir. 30 (41), 12306 (2014). doi:10.1021/la5032619.
- F. Zasada, W. Piskorz, S. Godlewski, et al., J. Phys. Chem. C. 115 (10), 4134 (2011). doi:10.1021/jp111014r.
- S. Begonja, L.A. García Rodenas, E.B. Borghi, et al., Colloids Surf., A. 403, 114 (2012). doi:10.1016/j.colsurfa.2012.03.064.
- I.A. Mudunkotuwa and V.H. Grassian, J. Am. Chem. Soc. 132 (42), 14986 (2010). doi:10.1021/ja106091q.
- J. Cheng and M. Sprik, J. Chem. Theory Comput. 6 (3), 880 (2010). doi:10.1021/ct100013q.
- G. Di Liberto, F. Maleki and G. Pacchioni, J. Phys. Chem. C. 126 (24), 10216 (2022). doi:10.1021/acs.jpcc.2c02289.
- J.M. Flitcroft, M. Molinari, N.A. Brincat, et al., J. Mater. Chem. A. 6 (24), 11362 (2018). doi:10.1039/C8TA02817F.