
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Formaldehyde is a highly reactive chemical that is usually sold and processed in the form of aqueous solutions, with methanol added for stability. In these solutions, formaldehyde reacts with the solvents to form a variety of reaction products, including oligomers. These chemical reactions can occur in the liquid and vapour phases and have a significant influence on the properties of formaldehyde-containing solutions. Of particular interest to industrial applications is the prediction of the vapour–liquid equilibria (VLE) in formaldehyde solutions, considering the chemical reactions. We use the SAFT-γ Mie group-contribution (GC) equation of state to obtain the fluid-phase behaviour of binary and ternary mixtures of formaldehyde with water and methanol. The oligomerisation reactions taking place in aqueous and methanolic solutions of formaldehyde are modelled implicitly using a physical approach, which is possible within the SAFT-γ Mie framework by adding association (reactive) sites that mediate the formation of the reaction products. Using this approach, the nature of the chemical speciation in formaldehyde + water, formaldehyde + methanol and formaldehyde + water + methanol mixtures is studied. A new group, CHO, characterising formaldehyde within the SAFT-γ Mie GC approach, is developed. Experimental data for the VLE in binary mixtures of formaldehyde + water and formaldehyde + methanol are used to obtain the optimal unlike interaction parameters between the corresponding SAFT-γ Mie groups. The newly developed parameters are used to predict the VLE of ternary formaldehyde + water + methanol mixtures for a wide range of temperatures and pressures, with excellent agreement to experimental data. Additionally, the SAFT-γ Mie approach is shown to provide accurate predictions of the distribution of reaction species (oligomers) in binary and ternary mixtures containing formaldehyde.
GRAPHICAL ABSTRACT
1. Introduction
Formaldehyde is an important industrial chemical. There are numerous applications of formaldehyde due to its versatility and ability to react with organic and inorganic compounds [Citation1]. In the pharmaceutical industry, formaldehyde is used as a raw material for the synthesis of various chemicals such as polyethylene glycol (PEG) and glycerol [Citation2–4]. Due to its high reactivity, however, formaldehyde can lead to the rapid degradation of pharmaceutical drug products during storage, significantly reducing the shelf-life of the drug [Citation5]. In addition to being present as an impurity in air, formaldehyde can be generated by some excipients (e.g., magnesium stearate and lactose) during storage [Citation6], negatively impacting the stability of drugs.
Pure monomeric formaldehyde is a colourless gas with a pungent smell, and is highly reactive. Due to its high reactivity, it is usually produced, stored, sold, and processed in the form of aqueous solutions. Methanol is sometimes added to enhance the stability of formaldehyde, to reduce the amount of water in solution, or to prevent the precipitation of polymers which may occur at low temperatures [Citation1,Citation2]. In aqueous and methanolic solutions, formaldehyde reacts with the solvents to form a variety of reaction products, which only exist in solution and cannot be isolated. The reaction of formaldehyde with water leads to the formation of methylene glycol (),
(1)
(1)
and poly(oxymethylene) glycols (
),
(2)
(2)
Similarly, the reaction between formaldehyde and methanol leads to the formation of hemiformal (HMF
),
(3)
(3)
and poly(oxymethylene) hemiformals (HMF
),
(4)
(4)
In formaldehyde + water + methanol mixtures, the four reactions take place simultaneously. These reactions can occur in the liquid and vapour phase, depending on the operating conditions used.
Formaldehyde is predominantly chemically bound to the solvents at all conditions in aqueous and methanolic solutions. Monomeric formaldehyde () makes up less than 0.04 wt%, even in highly concentrated solutions of formaldehyde [Citation2]. Therefore, the chemical reactions (Equation1
(1)
(1) )–(Equation4
(4)
(4) ) have a significant influence on the properties of the solutions and need to be considered in the thermodynamic modelling. Although, other chemical reactions may take place in formaldehyde mixtures with water and methanol, they have a negligible impact on the properties of the solutions and are not considered in the current work. The reader is referred to Walker [Citation2] and Maurer [Citation7] for more details of these reactions.
A large number of experimental measurements can be found for the vapour–liquid equilibria (VLE) of formaldehyde + water [Citation8–14], formaldehyde + methanol [Citation8,Citation14,Citation15], and of ternary formaldehyde + water + methanol mixtures [Citation7,Citation8,Citation13,Citation14,Citation16–18]. A common experimental method used by several authors [Citation7,Citation8,Citation16,Citation19] is the thin-film evaporator, in which a rotating coil is used to spread the liquid feed on the inner surface of a tube, surrounded by a heating jacket, causing the liquid to evaporate. After the separation of the liquid and vapour phases, the vapour phase is condensed. The two liquids are then collected in vials and analysed. To ensure that phase equilibrium is reached, a small evaporation ratio (the ratio of the volume of the condensed vapour to the volume of the unevaporated liquid) and long residence time is maintained in the experiments. To analyse the composition of the coexisting phases, gas chromatography is used to determine the water and/or methanol content, while the formaldehyde content is determined using the sodium sulfite method [Citation2]. At low temperatures (<320 K), the VLE of formaldehyde mixtures is significantly influenced by reaction kinetics and special care must be taken to ensure that chemical and phase equilibrium is attained. Hasse and Maurer [Citation8] have used a carrier-gas saturation technique to measure the VLE of formaldehyde + water, formaldehyde + methanol, and formaldehye + water + methanol mixtures. In this experimental method, an inert carrier-gas at a low flow rate is passed through, and is saturated with the formaldehyde liquid solution. During saturation, only a small fraction of the liquid evaporates, and the chemical equilibrium of the solution remains unaffected. The evaporated vapour is separated from the carrier gas in a cooling trap and is then condensed. Similar to the thin-film evaporator technique, the coexisting phases are analysed using the gas chromatography and sodium sulfite methods to determine their compositions.
For most industrial applications of formaldehyde, modelling the vapour–liquid equilibrium of aqueous and methanolic solutions of formaldehyde is of great importance. Extensive work on the thermodynamic modelling of the VLE of formaldehyde aqueous and methanolic solutions can be found in the literature. Perhaps one of the earliest modelling attempts of the VLE of formaldehyde mixtures is the physicochemical model presented by Maurer [Citation7] in 1986 to describe the VLE of binary mixtures of formaldehyde + water and formaldehyde + methanol using the universal functional activity coefficient (UNIFAC) [Citation20] model. In their model, experimental data were used to determine the necessary UNIFAC parameters, the thermodynamic equilibrium constants, and the vapour pressure of methylene glycol (or hemiformal) for each mixture. The performance of Maurer's model has been improved and its range of application extended, based on new experimental VLE data of formaldehyde mixtures [Citation8,Citation19,Citation21]. Specifically, an extension of the Maurer model to represent the ternary formaldehyde + water + methanol mixture was presented by Albert et al. [Citation16]. Using a different approach, Brandani et al. [Citation22] modelled the VLE of ternary mixtures of formaldehyde + water + methanol using the Wilson [Citation23] equation to calculate the activity coefficients of the three species. Brandani et al. [Citation24] subsequently modelled the isothermal VLE of formaldehyde + water and formaldehyde + methanol mixtures using the universal quasi-chemical (UNIQUAC) [Citation25,Citation26] equation to calculate the activity coefficients of the components in the mixtures. In addition to the unknown UNIQUAC parameters, the thermodynamic equilibrium constants for the reactions in the liquid phase were estimated from experimental VLE data. The Henry constant of formaldehyde in each solvent was also required for their model. This model was later extended to represent the ternary mixtures of formaldehyde + water + methanol by Brandani et al. [Citation27]. For the models discussed so far, the vapour phase was considered to be an ideal mixture of formaldehyde, water, methanol, methylene glycol, and hemiformal only. The concentration of higher oligomers produced by Reactions (Equation2(2)
(2) ) and (Equation4
(4)
(4) ) was assumed to be negligible in the vapour phase. The study by Brandani et al. [Citation28] was the first to account for dioxymethylene hemiformal (HMF
) in the vapour phase for a binary mixture of formaldehyde + methanol; they showed improved predictions, compared to previous studies, of the VLE of binary and ternary mixtures of formaldehyde, water, and methanol [Citation28]. This suggests that accounting for the oligomerisation reactions in the vapour phase, as well as in the liquid phase, can result in better models of the VLE of formaldehyde mixtures.
The equilibrium distribution of formaldehyde and its products with water (Reactions (Equation1(1)
(1) ) and (Equation2
(2)
(2) )) and methanol (Reactions (Equation3
(3)
(3) ) and (Equation4
(4)
(4) )) influences the thermodynamic phase equilibrium, chemical reaction kinetics, and transport properties. Therefore, quantitative information on the distribution of the reactions products in aqueous and methanolic solutions of formaldehyde is crucial for industrial purposes and has been the focus of several research studies over the years [Citation19,Citation21,Citation29–34]. Spectroscopic techniques can be used to determine the distribution and concentration of the species. UV/Vis spectroscopy can be used to study the formation of MG
and HMF
from monomeric formaldehyde (Reactions (Equation1
(1)
(1) ) and (Equation3
(3)
(3) ), respectively) [Citation29,Citation30]. More commonly, however, high-frequency nuclear-magnetic-resonance (NMR) spectroscopy is used to determine the formation of MG
and HMF
as well as the formation of the oligomers formed in Reactions (Equation2
(2)
(2) ) and (Equation4
(4)
(4) ). In NMR spectroscopy, separate peaks are assigned to the CH
groups in different MG
and HMF
in the mixture [Citation31]. The peak areas from the NMR analysis are converted to concentrations assuming they are proportional to the mole numbers of the CH
groups in solution. Multiple studies on the speciation analysis for the binary mixture of formaldehyde + water in the liquid phase have been undertaken using NMR spectroscopy [Citation19,Citation21,Citation32,Citation33]. Hahnenstein et al. [Citation32] studied the species distribution for the binary mixture of formaldehyde + methanol in the liquid phase using the NMR technique. Maiwald et al. [Citation34] later extended the work to investigate the distribution of species in the ternary mixture of formaldehyde + water + methanol in the liquid phase using NMR spectroscopy. One should note that, no peak could be assigned for monomeric formaldehyde in these NMR studies, due to its extremely low concentration in the liquid phase; hence, the true concentration of monomeric formaldehyde was considered to be negligible.
In an attempt to model the distribution of the species in a binary mixture of formaldehyde and water, Albert et al. [Citation21] used NMR spectroscopic data for formaldehyde + water mixtures [Citation32], in addition to experimental VLE data, to determine the reaction equilibrium constants (for the formation of the oligomers) and the unknown UNIFAC parameters in their work. Their model [Citation21] provided a reliable description of the formation of different MG in liquid mixtures of formaldehyde and water for a temperature range of 290–420 K. The extended model by Albert et al. [Citation16] resulted in good agreement with NMR speciation data of Hahnenstein et al. [Citation32] for the liquid mixture formaldehyde + methanol. Maiwald et al. [Citation34] used the model by Albert et al. [Citation16] to predict the distribution of the reaction products in a ternary liquid mixture of formaldehyde, water, and methanol. For ternary mixtures, this model [Citation16] provided only qualitative agreement with NMR experimental data [Citation34]. The most significant deviations were observed for the concentrations of MG
.
The literature discussed in the previous paragraphs on the modelling attempts for the VLE and the nature of speciation in formaldehyde mixtures follows a chemical approach to deal with the occurrence of chemical reactions and the formation of new species in each phase. In such methods, the reaction products are defined a priori and are treated explicitly within the thermodynamic model. An alternative to this methodology is the physical approach of dealing with reactions, in which the reaction products are treated implicitly and are considered to be aggregates of the reactants. In physical approaches, the reaction products do not need to be defined explicitly or known in advance, and their formation is driven by the presence of strong intermolecular interactions. It has been shown that physical approaches based on perturbation theories such as the statistical associating fluid theory (SAFT) [Citation35,Citation36] provide equivalent results to the chemical or quasi-chemical theories for the formation of hydrogen bonding between species in a mixture [Citation37]. In SAFT approaches, aggregates (reaction products) may form due to association interactions between the species, similar to the association approach used to model hydrogen bonding. The theory of chemical association within SAFT is based on the work of Wertheim [Citation38–41] in which the contribution to the Helmholtz free energy, due to association of a fluid composed of associating monomers, is evaluated. The short-range directional forces are accounted for by specifying off-center association sites on the monomers, which interact via a potential function. The parameters obtained in the physical theory to describe the association interaction between the reacting species can be related to the chemical reaction equilibrium constant in chemical theories [Citation37]. An advantage of physical approaches to modelling reactions over chemical approaches is that knowledge of the reaction equilibrium constants or the concentrations of the reaction products is not necessary for model development, which reduces the reliance on experimental data.
The physical approach to modelling reactions has been successfully implemented for carbon-capture processes within the SAFT framework [Citation42–45]. Mac Dowell et al. [Citation42] and Rodríguez et al. [Citation43] used the statistical associating fluid theory for potentials of variable range (SAFT-VR) [Citation46,Citation47], with square-well (SW) potentials, to describe the phase behaviour of reactive aqueous mixtures of carbon dioxide (CO) with alkanolamines. SAFT-VR SW is a homonuclear version of the SAFT theory in which intermolecular parameters are used to represent whole molecules rather than specific functional groups. An accurate representation of the complex reactions that occur in these mixtures was provided [Citation42,Citation43] by incorporating two association sites on the CO
molecule which can only interact with alkanolamines to form tightly bound aggregates, implicitly representing the formation of the main reaction products (carbamate and bicarbonate). Additionally, Rodríguez et al. [Citation43] studied the degree of speciation in ternary mixtures of CO
, monoethanolamine (MEA), and water, providing excellent agreement with experimental data of the true mole fraction of carbamate and bicarbonate for various CO
loadings in the mixture, without using any experimentally measured reaction equilibrium constants. To increase the predictive capability of these models, the SAFT-γ SW [Citation48,Citation49] and the SAFT-γ Mie [Citation50–54] group-contribution approaches have been used to model the phase behaviour of mixtures containing CO
with different amines and solvents, using a physical approach to represent the reactions [Citation44,Citation45]. These physical models delivered an accurate representation of the fluid-phase behaviour and chemical speciation in CO
-containing mixtures of relevance to CO
capture.
In the current work, the SAFT-γ Mie GC approach is used to predict the VLE in binary and ternary formaldehyde mixtures with water and methanol. In particular, we follow a physical approach to account for the oligomerisation reactions (Reactions (Equation1(1)
(1) )–(Equation4
(4)
(4) )) that occur in the liquid and vapour phases, as part of the thermodynamic modelling of the fluid-phase behaviour of these mixtures. This is done by adding a reactive association site on the group representing formaldehyde within the SAFT-γ Mie framework. In our model, the vapour phase is considered to be a mixture of real gases in which there is no limit on the length of oligomers that may form. Additionally, we investigate the nature of speciation in aqueous and methanolic mixtures of formaldehyde and predictively quantify the distribution of oligomers (reaction products) in various mixtures.
In the following section we present the thermodynamic modelling using the SAFT-γ Mie GC approach relevant to mixtures containing formaldehyde with water and methanol, including: a background on the SAFT-γ Mie theory; property calculations using the equation of state (EoS); the molecular models developed; and the parameter estimation strategy used within the SAFT-γ Mie GC approach. In Section 3, the phase equilibria calculations for pure formaldehyde, and its binary and ternary mixtures with water and methanol, are discussed. Following this, in Section 4, we present the methodology used to determine the distribution of reaction products in mixtures of formaldehyde, and the corresponding SAFT-γ Mie predictions of the speciation taking place in formaldehyde + water, formaldehyde + methanol, and formaldehyde + water + methanol mixtures. Final remarks are provided in Section 5.
2. Theory and thermodynamic modelling
2.1. SAFT-γ Mie theory
The theoretical background of the equation of state (EoS) is presented in detail in refs [Citation50–53] and a review of the latest developments in thermodynamic modelling using the SAFT-γ Mie EoS is presented in [Citation54]. Here we provide only a brief summary of the models.
The SAFT-γ Mie [Citation50–53] EoS is a group-contribution (GC) approach in which molecules are modelled as heteronuclear chains of fused-spherical segments with association sites, to account for short-range directional forces where appropriate. Within the SAFT-γ Mie framework, a group k is characterised by a number of spherical segments and a shape factor
which represents the contribution of each segment to the overall thermodynamic properties of the molecule considered. The interaction between two groups k and l is described using the Mie [Citation55] potential:
(5)
(5)
where
is the distance between the centers of the segments,
the segment diameter,
the depth of the potential well (dispersion energy), and
and
are the repulsive and attractive exponents of the segment-segment interaction, respectively. The prefactor
is a function of the Mie potential exponents,
(6)
(6)
which ensures the minimum of the interaction potential is
. The attractive exponent
is usually set to the London dispersion value of 6. Hydrogen bonding and strong polar interactions are treated through incorporating short-range square-well association sites on any given segment when appropriate. A segment k can have a number
of association site types, with
sites of each type
. The association interaction between square-well sites of type a on segment k and b on segment l is given by
(7)
(7)
where
is the distance between the centers of sites a and b,
is the association energy, and
is the cutoff range of the interaction between the two sites, which can be equivalently described in terms of the bonding volume
[Citation56]. Each site is positioned at a distance
or
from the center of the segment on which it is placed.
In summary, a group k is characterised by a set of like interaction parameters: the number of identical spherical segments; a shape factor
; the segment energy of interaction
; the segment diameter
; the exponents of the Mie potential
and
; and, where relevant, the parameters describing any site-site association interactions,
,
,
,
, …,
. The interactions between two groups k and l of different types are represented analogously by the corresponding unlike interaction parameters
,
,
,
,
, and
.
In the SAFT-γ Mie EoS the Helmoholtz free energy A of a mixture is written as a sum of terms, based on a perturbation approach, in which
(8)
(8)
where:
corresponds to the ideal gas free energy;
is the effect of the interaction of monomeric segments through Mie potentials;
is the contribution to the free energy of the formation of chains of molecules from the fused Mie segments; and
accounts for molecular association through short-range directional interactions. The first four terms describe the classical, non-electrolyte SAFT contributions [Citation50–53], and the presence of charged species is accounted for by the last two terms
and
[Citation57,Citation58].
Of particular importance to the current work is the association contribution term, which follows from the TPT1 expressions of Wertheim [Citation38–41,Citation56,Citation59] and is given by
(9)
(9)
where
is the number of components in the mixture,
is the number of groups,
is the mole fraction of species i, and
is the number of sites of type a on group k.
is the fraction of molecules of component i that are not bonded at a site of type a on group k, which can be obtained from the solution of the mass-action equations as [Citation41,Citation48,Citation56]
(10)
(10)
Here, ρ is the molecular density and
characterises the overall strength of the association between a site of type a on a group k of component i and a site of type b on a group l of component j. This is approximated as [Citation56]
(11)
(11)
where
and
is a temperature-density correlation for the association integral for a Lenard-Jones monomer [Citation52,Citation53].
2.2. Property calculations
The SAFT-γ Mie EoS, expressed in terms of the total Helmholtz free energy in Equation (Equation8(8)
(8) ), is a function of temperature T, volume V , and a vector N of the mole numbers of all the components in the mixture. Other properties can be calculated from standard thermodynamic relations and phase-equilibrium conditions [Citation60,Citation61].
The thermodynamic phase equilibrium of an isolated multi-phase mixture of i components can be obtained by imposing the equality of the temperature T, pressure P, and chemical potential of each component in each phase [Citation60]:
(12)
(12)
for all the α and β phases considered, to obtain the compositions of each phase. The pressure and chemical potential of component i can be calculated from the Helmholtz free energy as
(13)
(13)
and
(14)
(14)
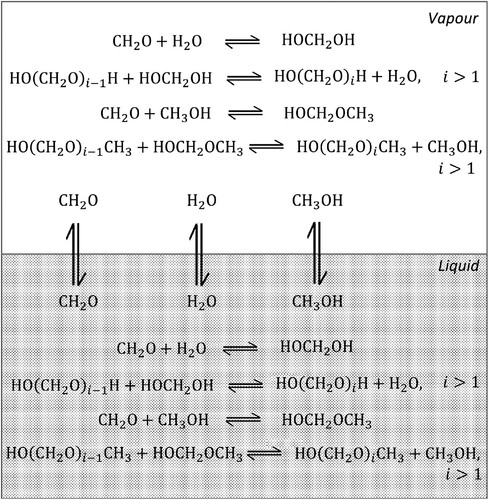
The relevant equilibria and chemical reactions for a mixture of formaldehyde, water, and methanol are shown in Figure . The liquid and vapour phases consist of methanol (
), water (
), and monomeric formaldehyde (
) as well as the reaction products of formaldehyde with these solvents: hemiformal (
), poly(oxymethylene) hemiformals (
), methylene glycol (
), and poly(oxymethylene) glycols (
). In our model, we account for the four chemical reactions given by Reactions (Equation1
(1)
(1) )–(Equation4
(4)
(4) ) in both the liquid and vapour phases.
Figure 1. Schematic representation of the vapour–liquid equilibria in a ternary mixture of formaldehyde + water + methanol. The liquid and vapour phases consist of methanol (), water (
) and monomeric formaldehyde (
). The reactions of formaldehyde with these solvents (Reactions (Equation1
(1)
(1) )–(Equation4
(4)
(4) )) are considered in the two phases, forming: hemiformal (
), poly(oxymethylene) hemiformals (
), methylene glycol (
), and poly(oxymethylene) glycols (
).
2.3. Molecular models
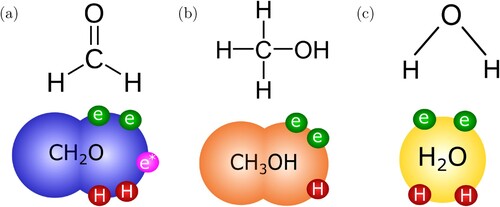
Using the SAFT-γ Mie GC approach, we need to characterise the groups that make up each of the compounds of interest. As formaldehyde, water, and methanol are relatively small molecules; they are each modelled as a single molecular group. The groups required for modelling these compounds are presented in Figure . These are: 1 × for formaldehyde; 1 ×
for methanol; and 1 ×
for water. Each group has two sites of type e which represent the two electron lone-pairs of the oxygen atom, and sites of type H corresponding to each of the hydrogen atoms (that may participate in hydrogen bonding), such that,
has one H site,
has two H sites, and
has two H sites. In our model, only sites of different type are allowed to interact:
for
and
.
Figure 2. Chemical structures of (a) formaldehyde, (b) methanol and (c) water considered in the current work, together with their corresponding SAFT-γ Mie representation. Each of the large coloured spheres corresponds to a different group and the small spheres represent the association sites on the groups: H (red), e (green), and e (purple).
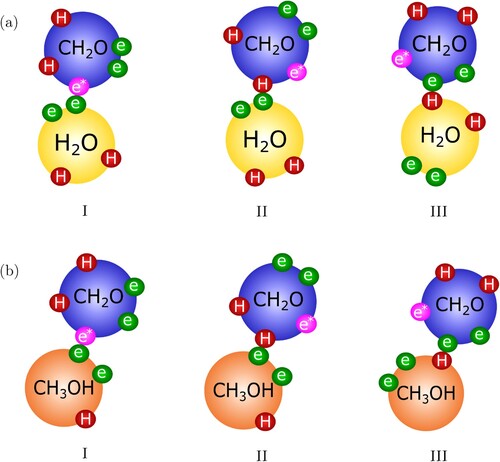
As discussed in Section 1, we use a physical approach in which the reaction products are considered to be aggregates of the reactants to model Reactions (Equation1(1)
(1) )–(Equation4
(4)
(4) ). This is possible within the SAFT-γ Mie framework by adding association (reactive) sites to mediate the formation of the reaction products. For a physical treatment of Reactions (Equation1
(1)
(1) )–(Equation4
(4)
(4) ), we add a reactive e
site to the
group which can only interact with the e sites on
and
:
. This reactive site is only active in a mixture but not in pure formaldehyde. Hence, the formation of methylene glycol (
) (Reaction (Equation1
(1)
(1) )) is mainly possible through the association between the e
site on
and the e sites on
(configuration I in Figure (a)), and the formation of hemiformal (
) (Reaction (Equation3
(3)
(3) )) mainly occurs through the association interaction between the e
site on
and the e sites on
(configuration I in Figure (b)). It is important to note that, because the e sites on
and
also associate with other site types, and not exclusively to the e
site on
,
and
may form due to other association interactions in our model.
may also form by an association interaction between the H site on
and the e site on
(configuration II in Figure (a)), or by the association interaction between the e site on
and the H site on
(configuration III in Figure (a)). Similarly,
can form by the association between the H site on
and the e site on
(configuration II in Figure (b)), or between the e site on
and the H site on
(configuration III in Figure (b)).
Figure 3. Configurations for the formation of (a) methylene glycol (MG) and (b) hemiformal (HMF
) mediated by e–H, and e–e
site-site bonds. e–e, e
–e
, H–H, or H–e
bonding is not allowed. We note that, for simplicity, the
and
groups are depicted as a single sphere; the reader is referred to Figure for the corresponding representation of the groups within the SAFT-γ Mie GC approach.
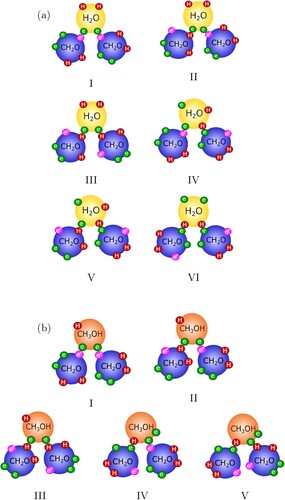
Our physical treatment, can also account for the formation of higher-order oligomers (Reactions (Equation2(2)
(2) ) and (Equation4
(4)
(4) )). For instance, there are six possible combinations of association interactions for the formation of dioxymethylene glycol (
) in our our model, these are shown in Figure (a). Likewise, dioxymethylene hemiformal (
) can form by one of the five association interaction combinations shown in Figure (b).
Figure 4. Configurations for the formation of (a) the dioxymethylene glycol (MG), and (b) the dioxymethylene hemiformal (HMF
), mediated by e–H, and e–e
site-site bonds. e–e, e
–e
, H–H, or H–e
bonding is not allowed. We note that, for simplicity, the
and
groups are depicted as a single sphere; the reader is referred to Figure for the corresponding representation of the groups within the SAFT-γ Mie GC approach.
The like and unlike SAFT-γ Mie group interactions for and
are obtained from previous work [Citation51–53]. In the current work, we expand the group-interactions matrix by estimating the SAFT-γ Mie parameters for the
group and its unlike interactions with
and
. The determination of the group-interaction parameters is carried out in a sequential manner. We first determine the like parameters for the
group, and these parameters are then used to obtain the unlike interaction parameters with the other groups. Details of the parameter-estimation strategy are outlined in the following section.
2.4. Parameter estimation
To estimate the SAFT-γ Mie parameters, the following least-squares objective function is minimised:
(15)
(15)
where Θ is the vector of model parameters,
is the number of property types,
is the number of experimental data points for property X,
is the ith measured value of property X,
is the corresponding value calculated by the SAFT-γ Mie EoS, and
refers to the the weight of data point i for property X. Here, we use equal weights for all the data points of all the properties:
. The parameter estimation is performed using the gPROMS [Citation62] software package.
To determine the accuracy of the theoretical description compared to the experimental data for a property X, the percentage absolute average deviation (%AAD)
(16)
(16)
and the absolute average deviation of (AAD)
(17)
(17)
are used.
The optimal SAFT-γ Mie parameters obtained for the group are presented in Tables , , and and are discussed in the following section. The unlike interaction parameters of
with
and
are provided in the tables, with a comparison of the SAFT-γ Mie predictions to experimental VLE data of binary mixtures of formaldehyde with water and methanol shown in Section 3.2. We also compare our SAFT-γ Mie predictions of the VLE for ternary formaldehyde + water + methanol mixtures with experimental data which were not used for parameter estimation in Section 3.3.
Table 1. SAFT-γ Mie parameters for water, methanol,' and formaldehyde.
Table 2. The percentage average absolute deviation %AAD between the SAFT-γ Mie calculations and experimental data of the saturated-liquid density , vapour pressure
, and vapourisation enthalpy
of formaldehyde.
Table 3. Group dispersion interaction energies and repulsive exponent
for use with the SAFT-γ Mie equation of state, taken from the references indicated in the last column.
Table 4. Group association energies and bonding volume
for use with the SAFT-γ Mie EoS.
indicates parameters obtained in the current work.
3. Phase equilibria
3.1. Pure compounds
To study the phase behaviour and speciation of formaldehyde solutions, the group needs to be characterised within the SAFT-γ Mie framework. In the current work, the
group (cf. Figure ) is modelled with two identical segments (
) and with three association site types (
): two sites of type e which represent the electron lone-pairs on the oxygen atom; two sites of type H corresponding to each of the hydrogen atoms; and one site of type e
as a reactive site. The attractive exponent of the Mie potential
is fixed to a value of 6.0. The remaining SAFT-γ Mie parameters for the
group are estimated using experimental data [Citation63,Citation64] of pure formaldehyde. Specifically, vapour pressure, saturated-liquid density, and enthalpy of vapourisation data are used. The set of optimal parameters can be found in Tables and , which also contain the previously published SAFT-γ Mie parameters for CH
OH and H
O [Citation51–53]. The SAFT-γ Mie calculations are compared to experimental data [Citation63,Citation64] in Figure , and the corresponding %AADs are presented in Table . As can be gleaned from the figure and %AADs, the model leads to a very good description of the thermodynamic properties of formaldehyde. Additionally, our model can accurately predict the critical temperature of formaldehyde (
) [Citation65] (cf. Figure (c)), although data in the critical region, above 0.9
, are not included in the parameter estimation.
Figure 5. (a) Vapour pressure, (b) saturated-liquid density, and (c) vaporisation enthalpy for pure formaldehyde. The curves correspond to the SAFT-γ Mie calculations and the symbols are experimental data [Citation63,Citation64]. The cross in (c) corresponds to the critical temperature of formaldehyde [Citation65].
![Figure 5. (a) Vapour pressure, (b) saturated-liquid density, and (c) vaporisation enthalpy for pure formaldehyde. The curves correspond to the SAFT-γ Mie calculations and the symbols are experimental data [Citation63,Citation64]. The cross in (c) corresponds to the critical temperature of formaldehyde [Citation65].](/cms/asset/51e65ae1-8d01-4b69-b61a-a2eea2ab77bc/tmph_a_2197712_f0005_oc.jpg)
3.2. Binary formaldehyde + water and formaldehyde + methanol mixtures
To describe the mixture of formaldehyde + water we use the model described in Section 2.3, following a physical approach to account for Reactions (Equation1(1)
(1) ) and (Equation2
(2)
(2) ) in the liquid and vapour phases. A total of eight unlike interactions need to be determined between the
and
groups, namely:
,
,
,
,
,
,
, and
. The repulsive exponent
is calculated with a combining rule. Experimental isobaric VLE data for formaldehyde + water mixtures [Citation10] over a pressure range of 0.013–0.053 MPa are used for the estimation of the remaining parameters using Equation (Equation15
(15)
(15) ). To reduce the number of parameters that have to be estimated, we consider a symmetric association scheme for the association interactions not involving the e
site:
, and
. The optimised parameters can be found in Tables and .
The SAFT-γ Mie calculations for the isobaric VLE of formaldehyde + water mixtures are in excellent agreement with the experimental data as shown in Figure (a,b), with corresponding %AADS for the bubble () and dew temperatures (
) of 0.12% and 0.12%, respectively (Table ). The phase boundaries are reported in the figures in terms of the ‘overall’ mole fraction of formaldehyde, always indicated by a (∼) in the current work,
; the ‘overall’ (
) and ‘true’ (
) mole fractions of formaldehyde can be related as discussed in Section 4.
Figure 6. (a) Isobaric () and (c) isothermal (
) phase diagrams featuring the vapour–liquid equilibria of formaldehyde + water binary mixtures, with an enlargement of the water-rich regions of the (b) isobaric and (d) isothermal phase diagrams. The filled symbols represent experimental data [Citation10] used in the parameter estimation, and the open symbols are experimental data (circles [Citation9] and squares [Citation66]) used for model validation only. The continuous curves represent calculations using SAFT-γ Mie. Error bars are shown for the experimental saturation points (squares [Citation66]) indicating the uncertainty associated with the measurements.
![Figure 6. (a) Isobaric (T−x~) and (c) isothermal (P−x~) phase diagrams featuring the vapour–liquid equilibria of formaldehyde + water binary mixtures, with an enlargement of the water-rich regions of the (b) isobaric and (d) isothermal phase diagrams. The filled symbols represent experimental data [Citation10] used in the parameter estimation, and the open symbols are experimental data (circles [Citation9] and squares [Citation66]) used for model validation only. The continuous curves represent calculations using SAFT-γ Mie. Error bars are shown for the experimental saturation points (squares [Citation66]) indicating the uncertainty associated with the measurements.](/cms/asset/1ec4d695-1ab6-41e8-bf91-0a56a02089ab/tmph_a_2197712_f0006_oc.jpg)
Table 5. The percentage average absolute deviation %AAD between the SAFT-γ Mie calculations and experimental data of the bubble and dew temperatures (,
) and pressures (
,
) for binary mixtures of formaldehyde + water (F + W) and formaldehyde + methanol (F + M).
We assess the validity of the model by predicting isothermal VLE data [Citation9] for temperatures and pressures not considered in the parameter estimation, and present a comparison between the SAFT-γ Mie predictions and experimental data in Figure (c,d). Very good agreement with the experimental data is apparent from the figures. The corresponding %AADs are 2.37% and 1.39% for the bubble () and dew (
) pressures, respectively (Table ). It is remarkable to see in Figure (b,d) that we are able to capture the azeotropes for all the isotherms and isobars considered for this binary mixture. Additionally, our predictions of the saturation points of formaldehyde are in close agreement with experimental measurements [Citation66], within experimental uncertainty, for all the temperatures and pressures assessed in Figure .
A similar approach is followed to model the phase behaviour of the binary mixture of formaldehyde and methanol, using a physical approach to account for Reactions (Equation3(3)
(3) ) and (Equation4
(4)
(4) ) in the liquid and vapour phases, as described in Section 2.3. Eight unlike interaction parameters are required for the
and
groups:
,
,
,
,
,
,
and
. The repulsive exponent
is calculated with a combining rules and a symmetric association scheme is considered for the association interactions between
and
not involving the reactive e
:
and
. These parameters are obtained following the parameter-estimation strategy outlined in Section 2.4, using experimental isobaric and isothermal VLE data for formaldehyde + methanol mixtures [Citation14,Citation15]. The estimated parameters are reported in Tables and .
Very good agreement is obtained between the SAFT-γ Mie calculations and experimental data for the VLE phase boundaries, as displayed in Figure . The azeotropes for all the isotherms and isobars are captured in close agreement to experimental data (see Figure (b,d)). The good agreement of our model with experimental data is also apparent from the %AADs presented in Table . Very low %AAD values of 0.67% and 1.94% are obtained for the and
, respectively, and slightly larger %AAD values are observed for the dew and bubble pressures with overall %AADs of 9.62% and 7.54%, respectively. The largest deviations for
and
are for the data points at the highest
of each isotherm, which correspond to the lowest pressures, where larger uncertainties can be expected in the experimental data. For example, the %AAD for
at
K and
is 27.83%, while the %AAD for the same property at the same temperature and
is 2.99%. It can also be noted from Figure (d) that, for the isotherms at 343.15 K and 353.15 K, the highest (at low
) and lowest (at high
) values for
(or
) differ by an order of magnitude, which explains the larger %AAD values obtained for the lower pressures. Therefore, it is the deviations at the largest
for each isotherm that contribute to higher overall %AADs for
and
. In the formaldehyde + water mixture, we did not observe large %AADs for
and
as the pressures at the lowest and highest
are of the same order of magnitude.
Figure 7. (a) Isobaric () and (c) isothermal (
) phase diagrams featuring the vapour–liquid equilibria of formaldehyde + methanol binary mixtures, with an enlargement of the methanol-rich regions of the (b) isobaric and (d) isothermal phase diagrams. The filled symbols represent experimental data (triangles [Citation14] and circles [Citation15]) used in the parameter estimation, and the open symbols are experimental data (circles [Citation15] and squares [Citation66]) used for model validation only. The continuous curves represent calculations using SAFT-γ Mie. Error bars are shown for the experimental saturation points (squares [Citation66]) indicating the uncertainty associated with the measurements.
![Figure 7. (a) Isobaric (T−x~) and (c) isothermal (P−x~) phase diagrams featuring the vapour–liquid equilibria of formaldehyde + methanol binary mixtures, with an enlargement of the methanol-rich regions of the (b) isobaric and (d) isothermal phase diagrams. The filled symbols represent experimental data (triangles [Citation14] and circles [Citation15]) used in the parameter estimation, and the open symbols are experimental data (circles [Citation15] and squares [Citation66]) used for model validation only. The continuous curves represent calculations using SAFT-γ Mie. Error bars are shown for the experimental saturation points (squares [Citation66]) indicating the uncertainty associated with the measurements.](/cms/asset/cde077d2-2a87-4da9-8517-6033eadd1966/tmph_a_2197712_f0007_oc.jpg)
There are, to the best of our knowledge, no experimental data for the VLE of binary formaldehyde + methanol (F + M) or formaldehyde + water (F + W) mixtures at high concentrations of formaldehyde ( for F + W, and
for F + M). Hence, the SAFT-γ Mie predictions in Figure (a,c) and Figure (a,c), provide insight of the VLE behaviour for these binary mixtures at high concentrations of formaldehyde at the given ranges of temperature and pressure.
In the last column of Table the value of for each association interaction, which is representative of the association strength (
) for a given association kernel
(cf. Equation (Equation11
(11)
(11) )), is presented at
K. It is evident that
of the interactions involving the reactive e
site on formaldehyde are several orders of magnitude larger than the other interactions. This suggests that the association interactions with the e
site on
are favorable when the corresponding sites are present in a mixture; the e site on
will preferentially associate with e
on
compared to the other sites on
,
, and
. Likewise, the e site on
will favor the association with e
on
compared to the other sites on
,
, and
. We note that the value of
is an order of magnitude larger than
, suggesting that, in a ternary formaldehyde + water + methanol mixture, the e
site on
is more likely to associate with the e site on
rather than the e site on
. The preferential association of
with
, as compared to
observed in our study is in agreement with findings by other authors [Citation2,Citation18] who suggest a higher affinity of formaldehyde for methanol as compared to water.
3.3. Ternary mixtures of formaldehyde
The VLE for ternary mixtures of formaldehyde + methanol + water is predicted using the SAFT-γ Mie parameters given in Tables , , and . No additional parameters are needed to carry out the calculations for the ternary mixture.
A wide range of experimental data for the VLE of the ternary mixture is used in the validation of the predictive capability of the SAFT-γ Mie EoS (cf. Table ). Generally, our SAFT-γ Mie model provides an accurate prediction of the VLE of the formaldehyde + methanol + water mixtures compared to all the experimental data sources reported in Table , with an overall %AAD of 3.78% for ,
,
, and
. The deviation between the calculations and experimental data, in terms of %AAD, for
and
is below 3%, which is excellent considering the wide range of pressures evaluated. The model developed also provides accurate predictions for the
and
, with %AADs typically below 8% and 11%, respectively. A larger deviation (31.5%) is observed between our SAFT-γ Mie calculations and experimental data of Hasse et al. [Citation8] for
of the ternary mixture. This larger deviation is in part due to the low values of pressure, such that small deviations from experimental data have a significant impact on the values of the relative %AAD. In this case, it is useful to consider the absolute deviations (AAD) for an overall assessment of the accuracy of the model. The low AAD for
(2.94
MPa) indicates a good agreement between our calculations and the experimental data of Hasse et al. [Citation8].
Table 6. The percentage average absolute deviation (%AAD) and the average absolute deviation (AAD) between the SAFT-γ Mie calculations and experimental data [Citation7,Citation8,Citation13,Citation14,Citation16–18] for the bubble and dew temperatures (,
) and pressures (
,
) of ternary mixtures of formaldehyde + water + methanol.
It is evident from Table that there is some variation in the experimental data for the same property by different authors. For example, the %AAD for between our calculations and the experimental data from Albert et al. [Citation16] and Kogan et al. [Citation17], who report data for a similar temperature range, is 1.92% and 7.60%, respectively. A similar discrepancy between these two data sources is observed for
. This highlights the uncertainty in the experimental data measurements, which is often not reported, and the importance of comparing predictive models to multiple sources of experimental data for appropriate validation.
In Figure we show a comparison between the SAFT-γ Mie VLE predictions of the ternary mixture and experimental data from Blazhin et al. [Citation14] at MPa and
MPa. For a given pressure and overall liquid-phase composition, the predictions of the equilibrium vapour-phase composition are found to be in good agreement with the experimental data. A closer agreement of the tie-lines connecting the liquid and vapour phases is evident for the isobaric data at 0.101330 MPa (Figure (b)) than at 0.026664 MPa (Figure (a)). This is also indicated by the lower AAD values in Table for the equilibrium vapour composition at 0.101330 MPa compared to 0.026664 MPa. In general, however, the model performs well in predicting the equilibrium vapour composition for all the components at 0.026664 MPa (AAD
) and 0.101330 MPa (AAD
), which is remarkable given that no ternary mixture data were included in the parameter estimation.
Figure 8. Isobaric phase diagrams featuring the vapour–liquid equilibria of formaldehyde + water + methanol ternary mixtures at (a) 0.026664 MPa (temperature range of 312–335 K) and (b) 0.101330 MPa (temperature range of 341–370 K). For a known overall composition of formaldehyde (), water (
), and methanol (
) in the liquid phase (red), the corresponding overall composition of formaldehyde (
), water (
), and methanol (
) in the vapour phase is predicted using SAFT-γ Mie (open green circles), and is compared to experimental data [Citation14] (filled green circles). The continuous and dashed blue tie-lines connect the overall liquid-phase composition to the experimental and predicted overall vapour-phase composition, respectively.
![Figure 8. Isobaric phase diagrams featuring the vapour–liquid equilibria of formaldehyde + water + methanol ternary mixtures at (a) 0.026664 MPa (temperature range of 312–335 K) and (b) 0.101330 MPa (temperature range of 341–370 K). For a known overall composition of formaldehyde (x~F), water (x~W), and methanol (x~M) in the liquid phase (red), the corresponding overall composition of formaldehyde (y~F), water (y~W), and methanol (y~M) in the vapour phase is predicted using SAFT-γ Mie (open green circles), and is compared to experimental data [Citation14] (filled green circles). The continuous and dashed blue tie-lines connect the overall liquid-phase composition to the experimental and predicted overall vapour-phase composition, respectively.](/cms/asset/afb89c95-e1bc-456a-9e80-26b3c045e6da/tmph_a_2197712_f0008_oc.jpg)
Table 7. The average absolute deviation (AAD) between the SAFT-γ Mie calculations and experimental data from refs [Citation14] and [Citation17] for the equilibrium mole fraction of formaldehyde (), water (
), and methanol (
) in the vapour phase of ternary mixtures of formaldehyde + water + methanol.
Similarly, in Figure the ternary-phase-diagram SAFT-γ Mie predictions of the overall vapour-phase composition in equilibrium with a preset overall liquid-phase composition, at 353 K, are shown compared to experimental data from Kogan et al. [Citation17]. We note that the experimental data by Kogan et al. [Citation17] were chosen for comparison in Figure as this data set gives the highest deviations for and
in Table (with the exception of the %AAD of
for the data reported by Hasse et al. [Citation8] as discussed previously). Additionally, the experimental data by Kogan et al. spans a wider formaldehyde composition range than that presented in the isobaric data of Blazhin et al., and can further validate the predictive capability of the SAFT-γ Mie GC approach for VLE predictions of the ternary mixture. Close agreement between our calculations and experimental data is evident for the isothermal data in Figure . It is apparent that the slopes and lengths of the predicted tie-lines in the ternary phase diagrams are in close agreement with the experimental tie-lines, particularly at low
, low
, and high
(bottom right vertex of the ternary plots). The performance of the SAFT-γ Mie model is also assessed in terms of the AAD for the equilibrium vapour-phase compositions of the three components at 333 K, 343 K, and 353 K, compared to the experimental data by Kogan et al. [Citation17], in Table . The overall deviations are low (AAD
), following a trend of increasing AAD values with decreasing temperature for all of the components.
Figure 9. Isothermal phase diagrams featuring the vapour–liquid equilibria of formaldehyde + water + methanol ternary mixtures at 353 K (pressure range 0.0449–0.161 MPa). For a known overall composition of formaldehyde (), water (
), and methanol (
) in the liquid phase (red circles), the corresponding overall composition of formaldehyde (
), water (
), and methanol (
) in the vapour phase is predicted using SAFT-γ Mie (open green circles), and is compared to experimental data [Citation17] (filled green circles). The continuous and dashed blue tie-lines connect the overall liquid-phase composition to the experimental and predicted overall vapour-phase composition, respectively. In (a)
,
, and
; (b)
,
, and
; and (c)
,
, and
.
![Figure 9. Isothermal phase diagrams featuring the vapour–liquid equilibria of formaldehyde + water + methanol ternary mixtures at 353 K (pressure range 0.0449–0.161 MPa). For a known overall composition of formaldehyde (x~F), water (x~W), and methanol (x~M) in the liquid phase (red circles), the corresponding overall composition of formaldehyde (y~F), water (y~W), and methanol (y~M) in the vapour phase is predicted using SAFT-γ Mie (open green circles), and is compared to experimental data [Citation17] (filled green circles). The continuous and dashed blue tie-lines connect the overall liquid-phase composition to the experimental and predicted overall vapour-phase composition, respectively. In (a) 0.0383≤x~F≤0.2237, 0.1317≤x~W≤0.7460, and 0.0303≤x~M≤0.8300; (b) 0.0621≤x~F≤0.3628, 0.1076≤x~W≤0.5975, and 0.0397≤x~M≤0.8303; and (c) 0.1138≤x~F≤0.5065, 0.0965≤x~W≤0.4275, and 0.0660≤x~M≤0.7897.](/cms/asset/73f9bee8-9687-42a9-9b6a-c85d5c386c42/tmph_a_2197712_f0009_oc.jpg)
The predictive capability of the SAFT-γ Mie GC approach is also validated in terms of the bubble-point predictions as shown in Figure , where we compare our calculations of the bubble pressure for a given temperature and equilibrium liquid-phase composition, to the experimental data of Brandani et al. [Citation18]. Excellent agreement between the calculated and measured is displayed for the compositions considered. Interestingly, the best agreement is obtained at the lowest temperature for each of the three liquid-phase compositions considered.
Figure 10. Constant-composition phase diagrams featuring the bubble point of formaldehyde + water + methanol ternary mixtures. The overall composition of formaldehyde (), water (
), and methanol (
) in each solution is (a)
,
,
; (b)
,
,
; and (c)
,
,
. The filled symbols the experimental data [Citation18] and the open symbols represent predictions using SAFT-γ Mie.
![Figure 10. Constant-composition phase diagrams featuring the bubble point of formaldehyde + water + methanol ternary mixtures. The overall composition of formaldehyde (x~F), water (x~W), and methanol (x~M) in each solution is (a) x~F=0.234, x~W=0.7642, x~M=0.0018; (b) x~F=0.2578, x~W=0.7352, x~M=0.007; and (c) x~F=0.3216, x~W=0.6735, x~M=0.0049. The filled symbols the experimental data [Citation18] and the open symbols represent predictions using SAFT-γ Mie.](/cms/asset/025f9380-e1d9-463e-a6b6-d0eb95ce1b21/tmph_a_2197712_f0010_oc.jpg)
4. Distribution of reaction species
In a ternary mixture of formaldehyde, methanol, and water, Reactions (Equation1(1)
(1) )–(Equation4
(4)
(4) ) take place simultaneously such that formaldehyde (F), methanol (M), water (W), methylene glycol (MG
), poly(oxy)methylene glycols (MG
), hemiformal (HMF
), and poly(oxy)methylene hemiformals (HMF
) are all present.
Suppose a ternary mixture is prepared by mixing moles of formaldehyde,
moles of methanol, and
moles of water. These numbers of moles are used to calculate the ‘overall’ mole fraction
of the mixture. However, by virtue of the oligomerisation reactions taking place (Reactions (Equation1
(1)
(1) )–(Equation4
(4)
(4) )), at a given T and P, the composition of the mixture is also specified by the ‘true’ number of moles of F, M, W, MG
, MG
, HMF
, and HMF
species which we denote here as,
,
,
,
,
,
, and
, respectively. The true and overall compositions of the components are related by mass balances:
(18)
(18)
(19)
(19)
(20)
(20)
(21)
(21)
and
(22)
(22)
Combining Equations (Equation21
(21)
(21) ) and (Equation22
(22)
(22) ) leads to
(23)
(23)
where
and
are the overall and true total mole numbers, respectively. Equations (Equation18
(18)
(18) )–(Equation23
(23)
(23) ) can be used to determine the compositions of the binary mixtures too, whereby: in mixtures of formaldehyde + water,
and,
; and in formaldehyde + methanol mixtures,
, and,
.
The distribution of formaldehyde into the different oligomer species produced in Reactions (Equation1(1)
(1) )–(Equation4
(4)
(4) ) in the liquid and vapour phases can be determined using the SAFT-γ Mie GC approach through the fraction
of molecules not bonded at a given site [Citation56,Citation59], which is calculated as part of the determination of the Helmholtz free energy in the SAFT EoS (Equation (Equation10
(10)
(10) )).
Given that is the fraction of molecules of component i not bonded at a site of type a on group k, (
) is the fraction of molecules of component i that are bonded at a site of type a on group k. For example, the proportion of formaldehyde molecules bonded at the e
association site on the
group is (
), and the corresponding true mole fraction is obtained by multiplying by the overall composition of formaldehyde:
. Purely statistical arguments are used to estimate the true mole fraction of each species, where the bonded fractions are treated as probabilities.
It is worth mentioning that for simplicity, in order to determine the distribution of reaction species in the current work, the formation of oligomers up to i = 2 only in Reactions (Equation2(2)
(2) ) and (Equation4
(4)
(4) ) is taken into account, although the SAFT-γ Mie molecular model presented for formaldehyde mixtures leads to the formation of longer oligomers. Hahnenstein et al. [Citation32] and Albert el al. [Citation19], have reported that the average number of
segments in
and
in aqueous and methanolic binary mixtures of formaldehyde is 2.1 and 1.1, respectively, at 293 K and an overall formaldehyde composition
. In aqueous solutions, the average number of
segments in
was found to decrease with increasing temperature, while the effect of temperature on the average number of
segments in
was seen to be negligible in methanolic solutions [Citation32]. Hence, in our analysis of the speciation occurring in formaldehyde mixtures, we assume that the true mole fraction of oligomers longer than
and
, in the liquid (
) and vapour (
) phases, is zero:
,
,
, and
for
.
In order to determine the composition of MG, MG
, HMF
, and HMF
, it is perhaps more convenient to express the mass balances in Equations (Equation18
(18)
(18) )–(Equation23
(23)
(23) ) in terms of the true,
, and overall,
mole fractions of the components. The corresponding mass balances in terms of the mole fractions of the components for a maximum chain length of i = 2 for MG
and HMF
, are given as
(24)
(24)
(25)
(25)
and
(26)
(26)
where
(27)
(27)
4.1. Binary mixtures
In the analysis of formaldehyde + water and formaldehyde + methanol binary mixtures, we use statistical arguments incorporating , calculated with the SAFT-γ Mie EoS, to determine the true mole fraction of MG
, MG
, HMF
, and HMF
from Equations (Equation24
(24)
(24) )–(Equation27
(27)
(27) ).
4.1.1. Methylene glycol (MG )
)
Reaction (Equation1(1)
(1) ) is a nucleophilic addition reaction in which water acts as a nucleophile, attracted by the partial positive charge of the carbon atom in the carbonyl group in formaldehyde, to form MG
; this is represented by configuration I in Figure (a). Here, however, we model all the possible association schemes that would lead to the formation of an MG
aggregate in the SAFT-γ Mie model (cf. Figure (a)). In order to determine the true mole fraction of MG
, we sum the probability of each configuration forming. For each MG
configuration, we multiply the probability that the two corresponding association sites in Figure (a) are bonded with the probability that no other sites on formaldehyde can bond to the bonded site-type on water.
Let us consider configuration I in Figure (a). MG forms when an e site on
and the e
site on
are bonded, the probability for which is given by
. Additionally, to ensure that no H sites on
bond to the free e site on
, we account for the probability that the two H sites on
are not bonded,
. Hence, the true mole fraction of MG
in configuration I,
, in Figure (a) is expressed as
(28)
(28)
Following the same approach for other configurations of MG
in Figure (a), the true overall mole fraction of MG
,
, in formaldehyde + water mixtures for configurations I, II, and III is approximated as
(29)
(29)
4.1.2. Dioxymethylene glycol (MG)
MG is produced via a condensation reaction between two MG
molecules according to Reaction (Equation2
(2)
(2) ) for
. Here, we model all the possible configurations for the formation of MG
(cf. Figure (a)). In order to approximate the true mole fraction of MG
,
, in formaldehyde + water binary mixtures, we sum the probability that each configuration would form. For example, for MG
to form in configuration I or II, the two e sites on
must be bonded, and the two H sites on
must be unbonded; to ensure that the bonding of e sites on
is to an H site and/or an e
site on
, we follow
(30)
(30) where
and
are the true mole fractions of MG
in configurations I and II, respectively. By multiplying
in Equation (Equation30
(30)
(30) ) we ensure that the
does not associate with other
or
groups via its H sites. For MG
to form in configuration III, IV, V, or VI we express the probabilities in terms of the sites bonded and not bonded on
:
(31)
(31)
We note that in each term in Equation (Equation31
(31)
(31) ) we trace whether the reactive e
site on
is bonded or not bonded. For configurations III–VI this is done by explicitly specifying whether the reactive e
site on
is bonded or not bonded. For configurations I and II, we assume that the two e sites on
are bonded to the e
site on
and not the H site on
, since the association strengths
as can be seen from Table .
In Table a breakdown of the bonded (tick) and not bonded (cross) sites on and
that are considered in the calculations of
and
in Equations (Equation29
(29)
(29) ) and (Equation31
(31)
(31) ) are shown.
Table 8. The association sites on formaldehyde, F, and water, W, which are bonded () and not-bonded (✗) for each configuration of MG
and MG
(cf. Figure (a) and (a)), in formaldehyde + water binary mixtures, as expressed in Equations (Equation29
(29)
(29) ) and (Equation31
(31)
(31) ), respectively.
4.1.3. Hemiformal (HMF)
Reaction (Equation3(3)
(3) ) is a nucleophilic addition reaction, similar to Reaction (Equation1
(1)
(1) ), in which methanol acts as a nucleophile, attracted by the partial positive charge of the carbon atom in the carbonyl group in formaldehyde, to form HMF
; this is represented by configuration I in Figure (b). As for MG
, we model all the possible configurations for the formation of HMF
in our SAFT-γ Mie model (cf. Figure (b)). The true mole fraction of HMF
,
, in formaldehyde + methanol mixtures can be determined by summing the probability that each configuration would form. For each HMF
configuration, we multiply the probability that the two corresponding association sites shown in Figure (b) are bonded. For example, in configuration I in Figure (b), HMF
will form when the e site on
and the e
site on
are bonded, the probability for which is given by
. Additionally, to ensure that no H sites on
bond to the free e site on
, we account for the probability that the two H sites on
are not bonded,
. Hence, the true mole fraction of HMF
forming in configuration I,
, in Figure (b) is expressed as
(32)
(32)
Therefore,
in formaldehyde + water mixtures is approximated as
(33)
(33)
4.1.4. Dioxymethylene hemiformal (HMF)
HMF is produced via a condensation reaction between two HMF
molecules according to Reaction (Equation4
(4)
(4) ) for
. Analogous to modelling MG
(Section 4.1.2), we consider all the possible configurations for the formation of HMF
(cf. Figure (b)). The true mole fraction of HMF
,
, in formaldehyde + methanol binary mixtures can be determined by considering the probability that HMF
will form via one of the configurations in Figure (b). For example, for HMF
to form in configuration I or II, the two e sites on
must be bonded and the H site on
must not be bonded, such that
(34)
(34)
where
and
are the true mole fractions of HMF
in configurations I and II, respectively. By specifying
we ensure that the
group does not associate with other
or
groups via the H site. For HMF
to form in configuration III, IV or V we express the probabilities in terms of the sites bonded and not bonded on
:
(35)
(35)
We note that in each term in Equation (Equation35
(35)
(35) ), we trace the bonding status of the reactive e
site on
. For configurations III–V this is done by explicitly specifying whether the reactive e
site on
is bonded or not-bonded. For configurations I and II, we assume that the two e sites on
are bonded to the e
site on
and not the H site on
, since the association strengths
as can be seen from Table .
In Table a breakdown of the bonded (tick) and not-bonded (cross) sites on and
that are considered in the calculations of
and
in Equations (Equation33
(33)
(33) ) and (Equation35
(35)
(35) ) are shown.
Table 9. The association sites on formaldehyde, F, and methanol, M, which are bonded () and not-bonded (✗) for each configuration of HMF
and HMF
(cf. Figure (b) and (b)), in formaldehyde + methanol mixtures, as expressed in Equations (Equation33
(33)
(33) ) and (Equation35
(35)
(35) ), respectively.
4.2. Ternary mixtures
In order to predict the distribution of the reaction species in ternary mixtures of formaldehyde + water + methanol, the component mass balances are used as well as information on the fraction of molecules not bonded from the SAFT-γ Mie EoS. The complexity of estimating the distribution of species increases for a ternary mixture due to the presence of an additional component in the solution which competes for the association sites on
. Therefore, expressions involving
of the components are more specific for ternary mixtures, compared to binary ones, where information on the sites bonded on the two components making up the oligomer are specified for each configuration; this is in contrast to specifying the sites bonded on only one of the components for binary mixtures (cf. Equations (Equation31
(31)
(31) ) and (Equation35
(35)
(35) )).
In the current work, and
are approximated using expressions of
then,
and
are calculated from the material balances given in Equations (Equation24
(24)
(24) )–(Equation27
(27)
(27) ).
4.2.1. MG
The true mole fraction of MG,
, in formaldehyde + water + methanol ternary mixtures is determined by the probability that MG
will form via one of the configurations in Figure (a). For example, for MG
to form in configuration I, the two e sites on
must be bonded and the two H sites on
must be unbonded, in addition to the e
sites on the two
molecules being bonded:
(36)
(36)
Following a similar approach to calculate the probability of the formation of the remaining MG
configurations, the true mole fraction of MG
is approximated by
(37)
(37)
By comparing the calculation of
in binary and ternary mixtures, given by Equations (Equation31
(31)
(31) ) and (Equation37
(37)
(37) ), respectively, we note that for
in ternary mixtures we use information of the sites bonded or unbonded for the two components (formaldehyde and water) in each term in Equation (Equation37
(37)
(37) ). This is in contrast to using the fraction of bonded sites for one of the components in each term in Equation (Equation31
(31)
(31) ) for formaldehyde + water binary mixtures. In ternary formaldehyde + water + methanol mixtures, the third component (methanol in this case) is competing with water to associate with formaldehyde, hence, we follow a more specific formulation in Equation (Equation37
(37)
(37) ) to ensure that the calculation is accounting for the formation of MG
only. The differences in the formulations of
in binary and ternary mixtures is also shown by comparing Tables and for MG
.
Table 10. The association sites on formaldehyde, F, water, W, and methanol, M, which are bonded () and not-bonded (✗) for each configuration of MG
and HMF
(cf. Figure (a,b)), in formaldehyde + water + methanol mixtures, as expressed in Equations (Equation37
(37)
(37) ) and (Equation39
(39)
(39) ), respectively.
4.2.2. HMF
The true mole fraction of HMF,
, in formaldehyde + water + methanol ternary mixtures is determined from the probability that HMF
will form via one of the configurations in Figure (b). Hence, we sum the probability that each configuration would form. For example, for HMF
to form in configuration I, the two e sites on
must be bonded, the H site on
must be unbonded, and the e
sites on the two
molecules should be bonded:
(38)
(38)
Following a similar approach to calculate the probability of the formation of the remaining HMF
configurations,
is approximated by
(39)
(39)
By comparing the calculation of
in binary and ternary mixtures, given by Equations (Equation35
(35)
(35) ) and (Equation39
(39)
(39) ), respectively, we note that for
in ternary mixtures we use information of the sites bonded or unbonded for the two components (formaldehdye and water) in each term in Equation (Equation39
(39)
(39) ). This is in contrast to using the fraction of bonded sites for one of the components in each term in Equation (Equation35
(35)
(35) ) for formaldehyde + methanol binary mixtures. In the ternary mixture, the third component (water in this case) is competing with methanol to associate with formaldehyde, hence, we follow a more specific formulation in Equation (Equation39
(39)
(39) ) to ensure that the calculation is accounting for the formation of HMF
only. The differences in the formulations of
in binary and ternary mixtures is also shown by comparing Tables and for HMF
.
4.2.3. MG and HMF
As mentioned previously, and
are approximated by solving the mass balances in Equations (Equation24
(24)
(24) )–(Equation27
(27)
(27) ) simultaneously, and using
and
obtained from Equations (Equation37
(37)
(37) ) and (Equation39
(39)
(39) ), respectively. However, to solve Equations (Equation24
(24)
(24) )–(Equation27
(27)
(27) ) the true mole fraction of formaldehyde,
, and the true mole fraction of methanol,
, or water,
, must be known.
Pure formaldehyde predominantly forms when the e site on CH
O is not bonded hence,
is approximated by
(40)
(40)
Additionally, methanol forms when none of the sites on the CH
OH group are bonded or, when all of the sites (the two e sites and one H site) on a CH
OH group associate with sites on other CH
OH groups. Consequently, the true mole fraction of methanol,
, can be approximated as
(41)
(41)
4.3. Speciation predictions
To determine the distribution of species in binary and ternary mixtures containing formaldehyde we follow the methodology outlined in Sections 4.1 and 4.2, and compare our predictions to experimental data to assess the validity of our approximations. The experimental data in the literature [Citation16,Citation21,Citation32,Citation34,Citation67] are reported for the liquid phase only. Hence, the predictions by our model are only shown for the liquid phase. It is important to note, however, that the same methodology is applicable for predicting the distribution of species for any phase at any P, T, x.
4.3.1. Binary mixtures
In formaldehyde + water mixtures, methylene glycol (MG) and poly(oxy)methylene glycols (MG
) are formed according to Reactions (Equation1
(1)
(1) ) and (Equation2
(2)
(2) ). The expressions given in Equations (Equation29
(29)
(29) ) and (Equation31
(31)
(31) ) are used to calculate
and
, respectively. Reasonable agreement between our predictions and experimental data by Albert et al. [Citation21] can be seen in Figure (a,b) at 338.15 K and 368.15 K, respectively. Small deviations, in terms of AAD, for
and
are obtained at both temperatures (Table ). This level of agreement is especially pleasing considering the SAFT-γ Mie results are fully predictive.
Figure 11. The true liquid mole fraction of MG and MG
for a given overall formaldehyde liquid-phase mole fraction in formaldehyde + water binary mixtures at 0.101 MPa and (a) 338.15 K, and (b) 368.15 K. The symbols represent experimental data [Citation21] and the continuous curves are SAFT-γ predictions. In (c) and (d), at 338.15 K and 368.15 K, respectively, the dashed curves are SAFT-γ predictions of the true mole fraction of the configurations of
(cf. Figure (a)). In (e) and (f), at 338.15 K and 368.15 K, respectively, dashed curves are SAFT-γ predictions of the true mole fraction of the configurations of
(cf. Figure (a)). The continuous curves in (c)–(f) are SAFT-γ predictions of the total true mole fraction of the species (the summation of the dashed curves).
![Figure 11. The true liquid mole fraction of MG1 and MG2 for a given overall formaldehyde liquid-phase mole fraction in formaldehyde + water binary mixtures at 0.101 MPa and (a) 338.15 K, and (b) 368.15 K. The symbols represent experimental data [Citation21] and the continuous curves are SAFT-γ predictions. In (c) and (d), at 338.15 K and 368.15 K, respectively, the dashed curves are SAFT-γ predictions of the true mole fraction of the configurations of MG1 (cf. Figure 3(a)). In (e) and (f), at 338.15 K and 368.15 K, respectively, dashed curves are SAFT-γ predictions of the true mole fraction of the configurations of MG2 (cf. Figure 4(a)). The continuous curves in (c)–(f) are SAFT-γ predictions of the total true mole fraction of the species (the summation of the dashed curves).](/cms/asset/dc300b44-1d8a-489b-8420-9dceeb06b25f/tmph_a_2197712_f0011_oc.jpg)
Table 11. The average absolute deviation (AAD) between the SAFT-γ Mie calculations and experimental data [Citation16,Citation21] for the true mole fraction of MG (
) and MG
(
), and of HMF
, (
) and HMF
(
) in binary mixtures of formaldehyde + water and formaldehyde + methanol, respectively.
Furthermore, the mole fractions of each of the configurations of MG (cf. Figure (a)) are presented in Figure (c,d). The total true mole fraction of MG
,
, is the sum of the mole fractions of each of the configurations. As can be seen, configuration III is found in highest concentration, followed by configuration I; the mole fraction of configuration II, in which the H site on
and the e site on
are bonded, is found to be negligible. Similarly, in Figure (e,f) the true mole fractions of each configuration of MG
are shown. It is evident from Figure (e,f) that configurations I, II, and IV, in which the e
site on at least one of the
groups is bonded to an e site on
, are present in the highest mole fractions. The mole fractions of configurations III, V, and VI, which do not involve the association of any e
on
, are found to be negligible at the two temperatures.
We note that the predictions for at 338.15 K (Figure (a,e)) suggest that
(instead of zero) for
; this is due to the numerical values of
and
which are found to be slightly different from 1 and 0, respectively (cf. Equation (Equation31
(31)
(31) )). The calculated value of
is accounted for in the determination of the deviation of the model from experimental data (where
for
) at 338.15 K (Table ). A similar behaviour can be observed at 368.15 K (Figure (b,f)). The corresponding calculated value of
for
is also considered in the calculation of AAD
at 368.15 K (Table ).
Considering the case of formaldehyde + methanol mixtures, hemiformal (HMF) and poly(oxy)methylene hemiformals (HMF
) are formed according to Reactions (Equation3
(3)
(3) ) and (Equation4
(4)
(4) ), respectively. In our approach, the true mole fraction of HMF
and HMF
can be approximated using Equations (Equation33
(33)
(33) ) and (Equation35
(35)
(35) ), respectively. Good agreement between our predictions and experimental data of Albert et al. [Citation16] is shown in Figure for
, which is also evident from the low AADs reported in Table . However, higher deviations between the SAFT-γ Mie calculations and experimental data for
are observed in Table .
Figure 12. The true liquid mole fraction of HMF and HMF
for a given overall formaldehyde liquid-phase composition in formaldehyde + methanol binary mixtures at 276 K and 0.101 MPa. The symbols correspond to experimental data [Citation16,Citation32], and the curves to SAFT-γ Mie predictions of the different aggregates as indicated in each figure.
![Figure 12. The true liquid mole fraction of HMF1 and HMF2 for a given overall formaldehyde liquid-phase composition in formaldehyde + methanol binary mixtures at 276 K and 0.101 MPa. The symbols correspond to experimental data [Citation16,Citation32], and the curves to SAFT-γ Mie predictions of the different aggregates as indicated in each figure.](/cms/asset/50464428-629c-48fb-b6e9-f1dd8bfd52df/tmph_a_2197712_f0012_oc.jpg)
The concentration of each of the configurations of HMF and HMF
shown in Figures (b) and (b) are presented in Figure together with the corresponding total true mole fractions at each
. For HMF
, configuration III is seen in the highest mole fraction, followed by configuration I; the mole fraction of configuration II is found to be negligible. We show in Figure (c), that for HMF
, configurations I, II, and IV have the highest mole fractions, with the remaining configurations having negligible mole fractions in this mixture.
We note that the model predictions for at 276 K (Figure (a,c)) suggest that
(instead of zero) for
; this is due to the numerical values of
and
which are found to be slightly different from 1 and 0, respectively (cf. Equation (Equation35
(35)
(35) )). This calculated value of
is accounted for in the determination of the deviation of the model from experimental data (where
for
) in terms of the AAD of
at 276 K (Table ).
4.3.2. Ternary mixtures
In the ternary (formaldehyde + water + methanol) system, methylene glycol MG, hemiformal HMF
, poly(oxy)methylene glycols MG
, and poly(oxy)methylene hemiformals HMF
are formed. The SAFT-γ Mie EoS can be used to predict the true mole fractions of MG
, MG
, HMF
, and HMF
,
,
,
and
, respectively. The methodology discussed in Section 4.2 and Equations (Equation24
(24)
(24) )–(Equation27
(27)
(27) ) are used to predict the distribution of reaction species in the current work.
In Figure a comparison of the SAFT-γ Mie predictions for the distribution of species is compared with the appropriate experimental data. Two experimental data sets [Citation34] (A and B) are considered. The experimental data were obtained using C NMR spectroscopy, and the difference between the two experimental data sets is a consequence of the NMR techniques and instruments used [Citation34]. Very good agreement between our calculations and experimental data can be seen. The corresponding AADs for all the mixtures are shown in Table . Particularly accurate predictions are obtained for
,
, and
with overall AADs
. However, an over-estimation of
can be seen in all the ternary mixtures of Figure which leads to a slightly higher AAD for
(Table ). This is, in part, due to the assumption used in our model, that the maximum oligomer length of methylene glycols that form, is MG
, such that
. The overestimation of
indicates that perhaps longer MG
oligomers have non-negligible mole fractions.
Figure 13. The true liquid-phase mole fraction ((a), (c), and (e)) of MG, MG
, HMF
and HMF
and ((b), (d), and (f)) of each of the configurations of MG
(cf. Figure (a)) and HMF
(cf. Figure (b)) in ternary mixtures of formaldehyde + water + methanol. In (a) and (b) the ternary mixture is at
K,
MPa and the overall composition is
,
, and
. In (c) and (d) the ternary mixture is at
K,
MPa and the overall composition is
,
, and
. In (e) and (f) the ternary mixture is at
K,
MPa and the overall composition of
,
, and
. The blue and red bars correspond to experimental data [Citation34], and the gray bars to SAFT-γ Mie predictions. The coloured bars in (b), (d), and (f) are SAFT-γ Mie predictions.
![Figure 13. The true liquid-phase mole fraction ((a), (c), and (e)) of MG1, MG2, HMF1 and HMF2 and ((b), (d), and (f)) of each of the configurations of MG2 (cf. Figure 4(a)) and HMF2 (cf. Figure 4(b)) in ternary mixtures of formaldehyde + water + methanol. In (a) and (b) the ternary mixture is at T=348 K, P=0.101 MPa and the overall composition is x~F=0.3047, x~W=0.4866, and x~M=0.2087. In (c) and (d) the ternary mixture is at T=348 K, P=0.101 MPa and the overall composition is x~F=0.3323, x~W=0.3684, and x~M=0.2933. In (e) and (f) the ternary mixture is at T=298 K, P=0.101 MPa and the overall composition of x~F=0.3047, x~W=0.4866, and x~M=0.2087. The blue and red bars correspond to experimental data [Citation34], and the gray bars to SAFT-γ Mie predictions. The coloured bars in (b), (d), and (f) are SAFT-γ Mie predictions.](/cms/asset/bdc36d0a-0056-41d3-a3c3-2bfbe92d829e/tmph_a_2197712_f0013_oc.jpg)
Table 12. The average absolute deviation (AAD) between the SAFT-γ Mie predictions and experimental data [Citation34] for the true mole fraction of MG (
), MG
(
), HMF
(
) and HMF
(
), in the liquid phase of formaldehyde + water + methanol ternary mixtures at
MPa, corresponding to Figure .
The true mole fraction of each configuration considered for MG and HMF
in the ternary mixtures calculated from the fraction of molecules not bonded at given sites from the SAFT-γ Mie EoS using Equations (Equation37
(37)
(37) ) and (Equation39
(39)
(39) ), respectively, are shown in Figure . It is apparent that for both in MG
and HMF
, configurations I, II, and IV, which involve the association of the e
site of
, are present at the highest proportions. For HMF
and MG
this result is consistent with the findings for binary mixtures, and is expected given the higher association strength of the e sites of
and
with the e
site on
(cf. Table ).
5. Conclusions
We have presented the development of SAFT-γ Mie group-contribution (GC) models to predict the thermodynamic fluid-phase equilibria in binary and ternary mixtures containing formaldehyde with water and methanol. In the modelling of the phase equilibria of these mixtures, we account for the oligomerisation reactions that occur in the liquid and vapour phases. We follow a physical approach to representing the reactions, in which the reaction products are treated implicitly and are considered to be aggregates of the reactants.
We develop the SAFT-γ Mie parameters for the new CHO group representing formaldehyde by using experimental data of the saturated-liquid density, vapour pressure, and enthalpy of vapourisation in pure formaldehyde for the parameter estimation. Previously published parameters for CH
OH and H
O are used in the current work. The unlike interaction parameters between CH
O and H
O, and between CH
O and CH
OH, are estimated from to experimental vapour–liquid equilibria (VLE) data of binary mixtures of formaldehyde + water and formaldehyde + methanol, respectively. The newly obtained parameters are validated by comparing the SAFT-γ Mie predictions of the VLE in binary mixtures containing formaldehyde to experimental data which are not included in the parameter estimation. Excellent agreement with experimental data is obtained, and the model can be used reliably to capture the azeotropes in the VLE profiles of these binary mixtures. The VLE in ternary mixtures of formaldehyde + water + methanol is predicted using the SAFT-γ Mie GC approach without additional parameters; close agreement to the experimental data is obtained with an overall %AAD of 3.78%.
In our model, the oligomerisation reactions of formaldehyde with water and methanol are considered to take place in the liquid and vapour phases. The vapour phase is treated as a non-ideal gas, with the thermodynamic properties calculated using the SAFT-γ Mie equation of state (EoS). The reactions taking place in the liquid and vapour phases are modelled using a reactive, e, site on the formaldehyde group (CH
O), to mediate the formation of reaction products. The reactive e
site is only allowed to associate with the e sites on the methanol (CH
OH) and water (H
O) groups, and it remains inactive in solutions of pure formaldehyde. Other association interactions, which represent the hydrogen bonding that can take place between the groups in a mixture, are considered in the usual manner within the SAFT-γ Mie GC approach.
The nature of speciation and distribution of reaction products in binary and ternary mixtures of formaldehyde with water and methanol is investigated using information of the fraction of molecules i not bonded at a given site a on group k, , obtained from the SAFT-γ Mie EoS. Statistical arguments are used to approximate the true mole fraction of methylene glycol (MG
), dioxymethylene glycol (MG
), hemiformal (HMF
), and dioxymethylene hemiformal (HMF
) in formaldehyde + water, formaldehyde + methanol, and formaldehyde + water + methanol mixtures, where the bonded fractions are treated as probabilities. Good agreement with experimental data is obtained. Using our methodology, it is possible to determine the configuration which is most likely to form for each oligomer. Interestingly, we find that the MG
and HMF
configurations involving the association of the reactive e
site of CH
O exhibit the highest mole fractions in the ternary mixtures.
The findings from our current work demonstrate the applicability of the SAFT-γ Mie GC approach in studying the complex mixtures containing formaldehyde, in particular, in understanding the phase behaviour and the nature of speciation in binary and ternary mixtures of formaldehyde with water and methanol, which can influence the chemical reaction kinetics and transport properties of these mixtures. This can be done without requiring experimental information for these mixtures. The predictive SAFT-γ Mie model developed in the current work is relevant to various industrial applications involving formaldehyde, and is particularly useful for enhancing the stability and extending the shelf-life of drugs in the pharmaceutical industry.
Data statement
Data underlying this article can be accessed on Zenodo at https://doi.org/10.5281/zenodo.7867883.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- R.N. Hader, R.D. Wallace and R.W. McKinney, Ind. Eng. Chem. Res. 44 (7), 1508–1518 (1952). doi:10.1021/ie50511a016
- J.F. Walker, Formaldehyde, American Chemical Society Monograph Series, 3rd ed. (Reinhold Publishing Corporation, London, 1964).
- X. Liu, S. Ahlgren, H.A.A.J. Korthout, L.F. Salomé-Abarca, L.M. Bayona, R. Verpoorte and Y.H. Choi, J. Chromatogr. A 1532, 198–207 (2018). doi:10.1016/j.chroma.2017.2.009
- H. Matsuda, K. Mori, M. Tomioka, N. Kariyasu, T. Fukami, K. Kurihara, K. Tochigi and K. Tomono, Fluid Phase Equilib. 406, 116–123 (2015). doi:10.1016/j.fluid.2015.07.032
- Y. Wu, J. Levons, A.S. Narang, K. Raghavan and V.M. Rao, AAPS PharmSciTech 12 (4), 1248–1263 (2011). doi:10.1208/s12249-011-9677-z
- M. Fujita, T. Ueda and T. Handa, Chem. Pharm. Bull. 57 (10), 1096–1099 (2009). doi:10.1248/cpb.57.1096
- G. Maurer, AIChE J. 32 (6), 932–948 (1986). doi:10.1002/(ISSN)1547-5905
- H. Hasse and G. Maurer, Fluid Phase Equilib. 64, 185–199 (1991). doi:10.1016/0378-3812(91)90013-W
- L.V. Kogan, Y.M. Blazhin, S.K. Ogorodnikov and V.V. Kafarov, J. Appl. Chem. USSR 50 (12), 2552–2556 (1977).
- Y.L. Bondarenko, I.I. Sabylin, M.A. Kharisov, K.I. Strel'nikov, O.D. Soboleva, Y.M. Blazhin and S.K. Ogorodnikov, Promst. Sint. Kauch. (8), 2–4 (1981).
- V. Brandani, G. Di Giacomo and P.U. Foscolo, Ind. Eng. Chem. Process Des. Dev. 19 (1), 179–185 (1980). doi:10.1021/i260073a031
- S.J. Green and R.E. Vener, Ind. Eng. Chem. 47 (1), 103–109 (1955). doi:10.1021/ie50541a037
- V.M. Olevskii and I.F. Golub'ev, Tr. Gos. Nauchno Issled. Proektn. Inst. Azotn. Promst. Prod. Org. Sin. 4, 36–59 (1954).
- Y.M. Blazhin, L.V. Kogan, L.K. Vagina, V.E. Pastor, A.I. Morozova and S.K. Ogorodnikov, J. Appl. Chem. USSR 49 (174), 167–170 (1976).
- L.V. Kogan and S.K. Ogorodnikov, J. Appl. Chem. USSR 53 (1), 98–102 (1980).
- M. Albert, B. Coto García, C. Kuhnert, R. Peschla and G. Maurer, AIChE J. 46 (8), 1676–1687 (2000). doi:10.1002/(ISSN)1547-5905
- L.V. Kogan and S.K. Ogorodnikov, J. Appl. Chem. USSR 53 (1), 102 (1980).
- V. Brandani and G. Di Giacomo, Fluid Phase Equilib. 24 (3), 307–333 (1985). doi:10.1016/0378-3812(85)85011-1
- M. Albert, I. Hahnenstein, H. Hasse and G. Maurer, AIChE J. 42 (6), 1741–1752 (1996). doi:10.1002/(ISSN)1547-5905
- A. Fredenslund, R.L. Jones and J.M. Prausnitz, AIChE J. 21 (6), 1086–1099 (1975). doi:10.1002/(ISSN)1547-5905
- M. Albert, B. Coto García, C. Kreiter and G. Maurer, AIChE J. 45 (9), 2024–2033 (1999). doi:10.1002/(ISSN)1547-5905
- V. Brandani, G. Di Giacomo and V. Mucciante, Quaderni Dell'ingegnere Chimico Italiano 23 (7–8), 1–3 (1987).
- G.M. Wilson, J. Am. Chem. Soc. 86 (2), 127–130 (1964). doi:10.1021/ja01056a002
- S. Brandani, V. Brandani and G. Di Giacomo, Ind. Eng. Chem. Res. 30 (2), 414–420 (1991). doi:10.1021/ie00050a020
- D.S. Abrams and J.M. Prausnitz, AIChE J. 21 (1), 116–128 (1975). doi:10.1002/(ISSN)1547-5905
- G. Maurer and J.M. Prausnitz, Fluid Phase Equilib. 2 (2), 91–99 (1978). doi:10.1016/0378-3812(78)85002-X
- S. Brandani, V. Brandani and G.D. Giacomo, Ind. Eng. Chem. Res. 31 (7), 1792–1798 (1992). doi:10.1021/ie00007a026
- S. Brandani, V. Brandani and I. Tarquini, Ind. Eng. Chem. Res. 37 (8), 3485–3489 (1998). doi:10.1021/ie980020y
- A.V. Rudnev, E.P. Kalyazin, K.S. Kalugin and G.V. Kovalev, Russ. J. Phys. Chem. 51 (10), 2603–2606 (1977).
- H.-Gg. Schecker and G. Schulz, Z. Phys. Chem. (N. F.). 65, 221–224 (1969). doi:10.1524/zpch.1969.65.1_4.221
- D.J. Le Botlan, B.G. Mechin and G.J. Martin, Anal. Chem. 55 (3), 587–591 (1983). doi:10.1021/ac00254a041
- I. Hahnenstein, H. Hasse, C.G. Kreiter and G. Maurer, Ind. Eng. Chem. 33 (4), 1022–1029 (1994). doi:10.1021/ie00028a033
- A.L. Balashov, S.M. Danov, A. Golovkin, V.L. Krasnov and A.N. Ponomarev, Russ. J. Appl. Chem. 69 (2), 190–192 (1996).
- M. Maiwald, H.H. Fischer, M. Ott, R. Peschla, C. Kuhnert, C.G. Kreiter, G. Maurer and H. Hasse, Ind. Eng. Chem. Res. 42 (2), 259–266 (2003). doi:10.1021/ie0203072
- W.G. Chapman, K.E. Gubbins, G. Jackson and M. Radosz, Fluid Phase Equilib. 52, 31–38 (1989). doi:10.1016/0378-3812(89)80308-5
- W.G. Chapman, K.E. Gubbins, G. Jackson and M. Radosz, Ind. Eng. Chem. Res. 29 (8), 1709–1721 (1990). doi:10.1021/ie00104a021
- I.G. Economou and M.D. Donohue, AIChE J. 37 (12), 1875–1894 (1991). doi:10.1002/(ISSN)1547-5905
- M.S. Wertheim, J. Stat. Phys. 35 (1), 19–34 (1984). doi:10.1007/BF01017362
- M.S. Wertheim, J. Stat. Phys. 35 (1), 35–47 (1984). doi:10.1007/BF01017363
- M.S. Wertheim, J. Stat. Phys. 42 (3), 459–476 (1986). doi:10.1007/BF01127721
- M.S. Wertheim, J. Stat. Phys. 42 (3), 477–492 (1986). doi:10.1007/BF01127722
- N. Mac Dowell, F. Llovell, C.S. Adjiman, G. Jackson and A. Galindo, Ind. Eng. Chem. Res. 49 (4), 1883–1899 (2010). doi:10.1021/ie901014t
- J. Rodríiguez, N. Mac Dowell, F. Llovell, C.S. Adjiman, G. Jackson and A. Galindo, Mol. Phys. 110 (11-12), 1325–1348 (2012). doi:10.1080/00268976.2012.665504
- A. Chremos, E. Forte, V. Papaioannou, A. Galindo, G. Jackson and C.S. Adjiman, Fluid Phase Equilib. 407, 280–297 (2016). doi:10.1016/j.fluid.2015.07.052
- F. Perdomo, S. Khalit, E. Graham, F. Tzirakis, A. Papadopoulos, I. Tsivintzelis, A. Papadopoulos, P. Seferlis, A. Galindo, G. Jakson and C. Adjiman, p. In preparation (2022).
- A. Gil-Villegas, A. Galindo, P.J. Whitehead, S.J. Mills, G. Jackson and A.N. Burgess, J. Chem. Phys. 106 (10), 4168–4186 (1997). doi:10.1063/1.473101
- A. Galindo, L.A. Davies, A. Gil-Villegas and G. Jackson, Mol. Phys. 93 (2), 241–252 (1998). doi:10.1080/00268979809482207
- A. Lymperiadis, C.S. Adjiman, A. Galindo and G. Jackson, J. Chem. Phys. 127 (23), 234903 (2007). doi:10.1063/1.2813894
- A. Lymperiadis, C.S. Adjiman, G. Jackson and A. Galindo, Fluid Phase Equilib. 274 (1–2), 85–104 (2008). doi:10.1016/j.fluid.2008.08.005
- V. Papaioannou, T. Lafitte, C. Avendaño, C.S. Adjiman, G. Jackson, E.A. Müller and A. Galindo, J. Chem. Phys. 140 (5), 054107 (2014). doi:10.1063/1.4851455
- S. Dufal, V. Papaioannou, M. Sadeqzadeh, T. Pogiatzis, A. Chremos, C.S. Adjiman, G. Jackson and A. Galindo, J. Chem. Eng. Data 59 (10), 3272–3288 (2014). doi:10.1021/je500248h
- S. Dufal, T. Lafitte, A.J. Haslam, A. Galindo, G.N.I. Clark, C. Vega and G. Jackson, Mol. Phys. 113 (9–10), 948–984 (2015). doi:10.1080/00268976.2015.1029027
- S. Dufal, T. Lafitte, A.J. Haslam, A. Galindo, G.N.I. Clark, C. Vega and G. Jackson, Mol. Phys. 116 (2), 283–285 (2018). doi:10.1080/00268976.2017.1402604
- A.J. Haslam, A. González-Pérez, S. Di Lecce, S.H. Khalit, F.A. Perdomo, S. Kournopoulos, M. Kohns, T. Lindeboom, M. Wehbe, S. Febra, G. Jackson, C.S. Adjiman and A. Galindo, J. Chem. Eng. Data 65 (12), 5862–5890 (2020). doi:10.1021/acs.jced.0c00746
- G. Mie, Ann. Phys. 316 (8), 657–697 (1903). doi:10.1002/(ISSN)1521-3889
- G. Jackson, W.G. Chapman and K.E. Gubbins, Mol. Phys. 65 (1), 1–31 (1988). doi:10.1080/00268978800100821
- G. Lazarou, Ph. D. thesis, Imperial College London, 2017.
- D.K. Eriksen, G. Lazarou, A. Galindo, G. Jackson, C.S. Adjiman and A.J. Haslam, Mol. Phys. 114 (18), 2724–2749 (2016). doi:10.1080/00268976.2016.1236221
- W.G. Chapman, G. Jackson and K.E. Gubbins, Mol. Phys. 65 (5), 1057–1079 (1988). doi:10.1080/00268978800101601
- M.L. Michelsen, and J.M. Mollerup, Fundamentals & Computational Aspects (Tie-Line Publications, Holte, 2007).
- J.M. Prausnitz, R.N. Lichtenthaler and E. Gomes de Azevedo, Molecular Thermodynamics of Fluid-phase Equilibria, 3rd ed. (Prentice-Hall Inc., New Jersey, 1998).
- gPROMS v. 6.0.4, (PSE Ltd., London, 2019).
- G. Liessmann, W. Schmidt and S. Reiffarth, Recommended Thermophys Data 1 (1995).
- R. Spence and W. Wild, J. Chem. Soc. (Resumed), 506–509 (1935). doi:10.1039/jr9350000506
- H.R. Gerberich and G.C. Seaman, Kirk-Othmer Encyclopedia of Chemical Technology (American Cancer Society, Hoboken, NJ, USA, 2013).
- K. Kroenlein, C.D. Muzny, A.F. Kazakov, V. Diky, R.D. Chirico, J.W. Magee, I. Abdulagatov and M. Frenkel, NIST/TRC Web Thermo Tables (WTT) NIST Standard Reference Subscription Database 2-Lite Edition Version 2.
- M. Ott, H.H. Fischer, M. Maiwald, K. Albert and H. Hasse, Chem. Eng. Process. Process Intensif. 44 (6), 653–660 (2005). doi:10.1016/j.cep.2003.07.004