
Abstract
The search for the chemical structures that account for diffuse interstellar bands remains the main challenge in astrochemistry. It relies on experimental verification of structures obtained from stable isomers in laboratory conditions. The actual interstellar medium, however, is exposed to much harsher conditions. Herein we demonstrate the isomerisation of small, doubly charged polyaromatic hydrocarbon phenanthrene obtained by electron ionisation at relatively high electron energies. We performed electronic and vibrational tagging photodissociation spectroscopy measurements. Our results show that phenanthrene dication has a broad visible spectrum, making it unsuitable for DIB search. We also detected another C14H102+ isomer, originating from the isomerisation of phenanthrene dication during the ionisation process. Hole-burning spectroscopy experiments provided the IR spectrum of an isomer which could be assigned based on DFT calculations as a fully conjugated system representing the global minimum on the C14H102+ potential energy surface.
GRAPHICAL ABSTRACT

Introduction
In 1919 Mary Lea Heger, studying the interstellar sodium lines, observed two spectroscopic features associated with the interstellar medium (ISM) [Citation1]. This discovery has established a century-long spectroscopic research that identified more than 500 diffuse interstellar bands (DIBs) associated with the absorption of the ISM. However, the chemical composition of the ISM became the most challenging mystery in astrochemistry. The key to this mystery relies on constant improvement of experimental methods allowing for spectroscopic studies of candidate structures in laboratory conditions close to the ones in space, such as high vacuum and low temperature. The method of choice for the precise identification of charged DIB carriers is the cryogenic electronic ion spectroscopy, that intrinsically implies these parameters. The modern cryogenic ion spectroscopy started with the development of linear multipole in traps [Citation2] and is a method of choice for characterisation of ion properties, e.g. 3D structure and absorption [Citation3–11]. Numerous polycarbon molecules and ions have been proposed to account for DIBs [Citation12,Citation13]. However, only one ion, namely ionic fullerene C60+, has been experimentally confirmed so far to be a DIB carrier [Citation14–17].
Polycyclic aromatic hydrocarbons (PAH) of various sizes and charge states are another type of potential DIB carriers [Citation18]. The interest in these species is also supported by the observation of aromatic infared bands (AIB) in many astrophysical objects [Citation19–21]. These bands are suggested to stem from the PAH emission in mid-IR range [Citation22–24]. Among ionic PAHs, singly charged species have been dominantly considered [Citation25–34]. However, multiply charged PAHs can also be important candidates [Citation35]. The conditions in space may be harsh; the impact of high-energy electrons or radiation might result in higher charge states. Another effect of harsher conditions might be the isomerisation of ions and molecules to exotic candidate structures [Citation36].
This report presents electronic and vibrational spectroscopy of doubly charged PAH phenanthrene (Phen2+). We recorded the visible (Vis) and infrared (IR) spectra of the He-Phen2+ complexes at 3.4 K using tagging photodissociation methods in a cold ion trap and tabletop radiation sources. We show that electron ionisation of phenanthrene at 160 eV energy leads to the expected phenanthrene dication and another isomer lower in energy based on DFT calculations.
Experimental
Tagging photodissociation ion spectroscopy
The ions were generated using an electron ionisation source in a homebuilt instrument equipped with a cold ion trap for spectroscopic measurements [Citation37]. The ions of interest were mass-selected by a quadrupole mass filter, guided into a cold ion trap (∼3.4 K) by an octopule ion guide, and trapped and thermalised in collisions with helium buffer gas.
The duration of each cycle is 1s (see Scheme S1). It begins with filling the trap with ions for 200 ms. At the same time, the trap is filled with helium buffer gas that is injected by a piezo valve in several pulses of 0.2 ms separated by a 20 ms delay. The cold ions formed complexes with helium (He)n-Phen2+ resulting in approximately 40% of the trapped ions being tagged with helium atoms. In order to avoid the influence of secondary He clusters on the spectrum the conditions were tuned so that only singly tagged complex He-Phen2+ is observed and used for monitoring Vis or IR photon absorption. After the irradiation, the ions were extracted from the trap, mass-analyzed by a second quadrupole mass filter, and detected with a Daly-type detector working in the counting mode. The ion extraction time was 5 ms.
Electronic spectra were measured by a supercontinuum laser (NKT Photonics, SuperK Extreme EXB-6, seed laser 1065.7 nm, 77.945 MHz). The total output power is 600 mW in the vis range (430–530 nm: 1.1 mW/nm, 600–680 nm: 1.7 mW/nm). Using the acusto-optic tunable filter (AOTF) SuperK Select VIS/NIR AOTF the output was varied between 420 and 650 nm. Over this range, the FWHM varies from 1.8 nm to 8.5 nm. Vibrational spectra were measured by one of two pulsed tunable OPO/OPA systems (Laser Vision, 10 Hz repetition rate, resolution ∼1.6 cm−1 FWHM) operating in the far- and mid-IR range. The OPOs were pumped by a seeded Nd:YAG laser (Surelite EX from Continuum). The photon absorption at each wavelength was monitored as a depletion of the number of the He-Phen2+ complexes. The spectrum is plotted as 1-Ni/N0, where Ni and N0 were measured in alternating cycles (1 Hz) with the laser radiation on and off, respectively.
Two lasers, pump and probe, were used simultaneously during the hole-burning spectroscopy experiments [Citation38]. While the probe IR radiation was employed as described above, the pump Vis or far-IR radiation was used to destroy complexes that absorb at a selected wavelength. The resulting ion ensemble only contained clusters of isomer(s) that did not absorb the pump laser radiation (Scheme S2).
Calculations
The theoretical study has been performed at the density functional theory (DFT) level using the B3LYP functional and 6-311 + G(2d,p) basis set [Citation39,Citation40] as implemented in Gaussian 16 [Citation41]. The various isomers were pre-optimised at the B3LYP/6-31G** level. The infrared spectra were calculated for the optimised minima using harmonic approximation and an anharmonic GVPT2 approach. The harmonic IR spectra were scaled by 0.982 in the 900 cm−1–1700cm−1 range and by 0.965 in the 2700 cm−1–3300 cm−1 range [Citation42].
Results
Photodissociation spectroscopy
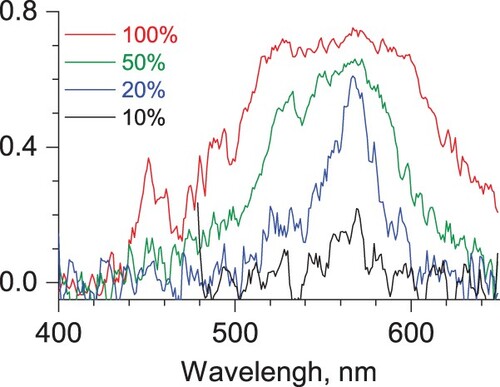
We employed electronic and vibrational tagging photodissociation spectroscopy to characterise doubly charged phenanthrene complexes with a He atom. Figure shows the visible tagging photodissociation spectrum of the He-Phen2+ dications generated from neutral phenanthrene and recorded at various power levels to monitor the power broadening of the absorption spectrum.
Figure 1. Electronic tagging photodissociation spectra of He-Phen2+ recorded at different levels of laser power. The Y-axis shows the absolute depletion value, 1-Ni/N0.
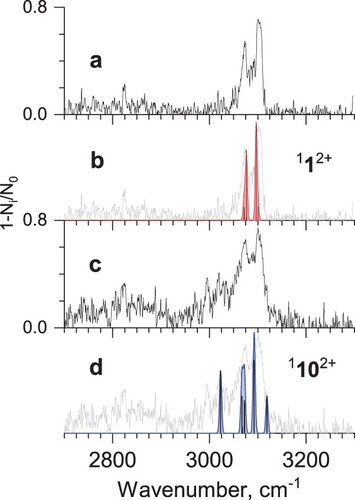
Vibrational spectra of He-Phen2+ complexes recorded by tagging photodissociation ion spectroscopy in the far- and mid-IR range are shown in Figure a and Figure a, respectively. Previously reported vibrational spectrum of Phen2+ in far-IR was measured using Ne atoms as tags and a free electron laser (FEL) as a light source [Citation43]. The spectra are similar (see Figure S1), but the comparison shows that using a lighter tag (He) and lower intensity radiation with a better resolution (tabletop IR OPO) results in slightly better-resolved vibrational bands. In addition, the He tag potentially causes less distortion to the gas-phase vibrational frequencies, compared to Ne, thus providing a more accurate benchmark for theoretical calculations [Citation37,Citation44–46].
Figure 2. a) Far-IR tagging photodissociation spectrum of He-Phen2+; b) calculated vibrational spectrum of phenanthrene dication 112+; c) Vis-IR hole-burning tagging photodissociation spectrum of He-Phen2+, recorded with Vis excitation fixed at 570 nm; d) calculated vibrational spectrum of isomer 1102+ (see Figure ).
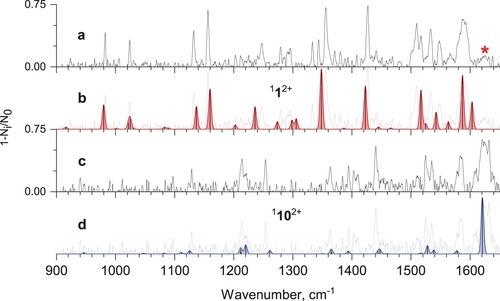
Figure 3. a) Mid-IR tagging photodissociation spectrum of He-Phen2+; b) calculated vibrational spectrum of phenanthrene dication 112+; c) IR-IR hole-burning tagging photodissociation spectrum of He-Phen2+, recorded with pump IR excitation fixed at 1427 cm−1; d) calculated vibrational spectrum of isomer 1102+ (see Figure ).
Surprisingly, the IR spectrum in Figure a shows a low-intensity band at 1625 cm−1 (marked with a red asterisk) that was not observed in the previous study and is not predicted by harmonic calculations. The band could correspond to either a combination or an overtone band or stem from an isomeric dication(s). To test the latter hypothesis, we employed Vis-IR hole-burning tagging photodissociation spectroscopy. This experiment is similar to the regular IR spectroscopic measurement, except that the ion ensemble was continuously irradiated by visible light fixed at 570 nm. The chosen wavelength corresponds to the maximum absorption of He-Phen2+ (Figure ). The resulting hole-burning spectrum is shown in Figure c. It is clear that, given almost full saturation of the pump signal, all vibrational transitions initially assigned to the phenanthrene dication disappear from the spectrum, hence the band at 1625 cm−1 cannot be attributed to the combination band or an overtone vibration. This result confirms that phenanthrene dications can isomerise during the ionisation process with high-energy electrons. The relative abundance of the minor isomer is around 20% based on the intensities of the bands at 1590 and 1625 cm−1.
Isolation of the minor isomer's mid-IR spectrum required modified hole-burning experiments because the visible and mid-IR laser beams enter the ion trap from the same side. Therefore, we used counter-propagating far-IR laser radiation at 1427 cm−1 as a pump. As a result of insufficient laser power, not all phenanthrene dication clusters might be depleted; hence the resulting hole-burning spectrum, shown in Figure c, might still contain features from both phenanthrene dication and its isomers.
DFT calculations
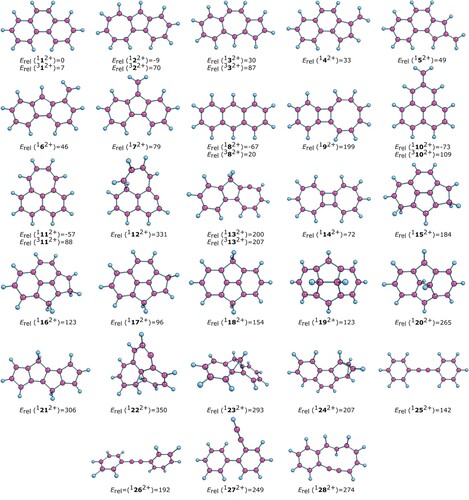
To identify the new C14H102+ isomer, we have explored various possible structures in the singlet and triplet states (Figure ). The calculated vibrational spectrum of Phen2+ in the ground electronic state, 112+, is shown in Figure b and Figure b (far- and mid-IR ranges, respectively). The artificial broadening of the lines to 5 cm−1 at FWHM was applied.
Figure 4. Geometries and relative energies in kJ mol−1 of various C14H102+ isomers. The energies are given at 0 K (including zero-point vibrational energy) relative to the energy of phenanthrene dication in the singlet ground state (112+). Note that 1252+ and 1262+ have -C≡C- motif between the phenyl rings appearing as one long connection and 1282+ has -C≡C- as a part of the macrocycle.
Discussion
Given the harsh conditions in the ISM, small, doubly charged PAHs might be relevant candidates to account for DIBs. Anthracene dication, measured previously at identical conditions, was shown to have a well-resolved electronic spectrum, which makes it potentially interesting as a candidate for the DIB search [Citation42]. The absorption of its isomeric phenanthrene dication lies in the same spectral range (Figure ). However, in striking odds with this closely related ion, the visible spectrum of phenanthrene dication is broad and unstructured even at low laser power FWHM ∼30 nm @20% power level (blue line), excluding it from the DIBs candidates in this spectral range. Possible reasons for the broad spectrum could be either a short excited state lifetime and/or higher Frank-Condon factors for vibronic transitions.
On the other hand, the vibrational spectrum of phenanthrene dication reveals intriguing features. An excellent match between theory and experiment (Figure and Figure b) together with consistency with data on Ne-Phen2+ reported previously [Citation43] (Figure S1) allow for an unambiguous identification of the vibrational transitions in our spectrum, associated with the phenanthrene dication in a singlet state, a structure denoted as 112+ in Figure .
In addition, the hole-burning experiments allowed us to isolate well-resolved vibrational transitions in the far-IR spectral range associated with a new isomer (Figure c). These isomeric ion does not absorb visible light at 570 nm used for the hole-burning experiments. This fact allows us to rule out the possibility that the unknown isomer is anthracene dication because it has strong absorption in this region [Citation42]. Furthermore, the most distinguishing feature, namely the most intense vibrational band at 1625 cm−1, is not present in the reported spectra of anthracene dication [Citation42,Citation43].
We suggest that this isomer is formed during the ionisation process. Isomerisation during the ionisation has been previously reported also for benzene dication [Citation47]. The resulting ion mixture is rapidly cooled in the ion trap, preventing further relaxation to more stable forms and allowing us to detect them spectroscopically. To identify the new C14H102+ isomer, we have explored 28 possible structures in the singlet state and 6 in the triplet state (Figure ). The triplet state of phenanthrene dication (312+) lies only 7 kJ mol−1 higher in energy than the singlet ground state (112+). The previously studied anthracene dication (182+) lies 67 kJ mol−1 lower in energy than the phenanthrene dication. The triplet state of anthracene dication lies 87 kJ mol−1 above its ground state.
For possible isomerisation, we have considered structures formed by a ring expansion or/and ring-contraction first. Such rearrangements lead to tricyclic isomers with the azulene sub-structure (122+ and 132+) and exo-methylene group (142+ - 172+). Isomer 122+ is more stable than the original phenanthrene dication, but the other isomers lie higher in energy than 112+.
Formation of other tricyclic structures would require several rearrangements or H-migration steps. Some possible isomers are shown in the second row of Figure . Next to the already mentioned anthracene dication, we found two other low-energy lying isomers, 1102+ and 1112+. These isomers have a different fusion of the rings, making the central motif of the phenalene isomer with an exo-methylene group (1102+) or enabling two six-membered rings and one seven-membered ring (1112+). 1102+ lies 73 kJ mol−1 lower in energy than 112+ and represents the lowest-lying structure in the calculation survey of the C14H102+ potential energy surface.
We have further explored ring fusions towards the structures with four rings (isomers 1152+ - 1222+). To our surprise, this structural modification did not yield any low-energy lying isomer. The most stable isomer in this category, 1172+, lies 96 kJ mol−1 higher in energy than 112+. We also explored the stability of isomers with one ring rearranged to a pyramidal structure (152+ and 162+). It is well known for benzene dication that the most stable structure corresponds to the pyramidal isomer [Citation47–49]. However, the polycyclic structure of C14H102+ does not favour the formation of a symmetrical pyramidal arrangement. The converged geometries corresponding to the pyramidal arrangement do not profit from creating a symmetrical pyramid with a five-membered base and a C–H group in the apex (1232+ and 1242+). As a result, these structures lie very high in energy. Finally, we explored isomers containing formally a C–C triple bond (-C≡C- in 1252+ - 1282+). All these isomers lie very high in energy; therefore, they are also unlikely candidates for our unknown isomer in the experimental data.
Figure compares the unknown isomer's experimental spectrum with the calculated harmonic ones for the most stable C14H102+ isomers (1102+, 1112+, 122+, 312+,382+,132+,142+, the remaining spectra are shown in Figure S2). We did not include the spectrum of anthracene dication in the singlet state (182+), because we could exclude its contribution based on the previously measured experimental spectrum [Citation42].
Figure 5. Harmonic IR spectra of the low-energy isomers of C14H102+.
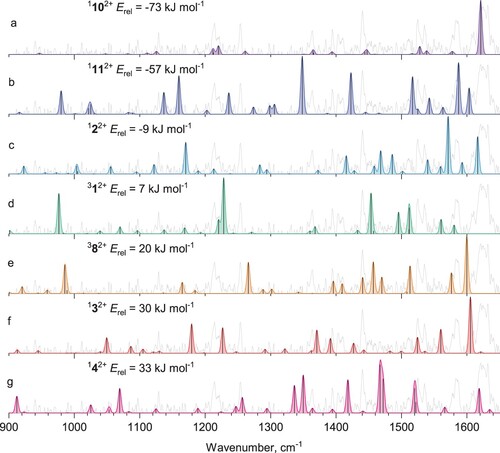
Among the other explored structures, we focused on the ones that could account for the strong, highest frequency band. Among the optimised structures, a vibrational transition with the blue-most frequency belongs to the most stable isomer in the singlet state 1102+, which lies 73 kJ mol−1 lower in energy than phenanthrene dication. The harmonic IR spectrum of this isomer is shown in Figure d and Figure d (far-and mid-IR ranges, respectively). The far-IR range shows an excellent match with the experimental spectrum.
As was mentioned above, the hole-burning spectrum of He-Phen2+ in the mid-IR range is likely to contain some contribution of the phenanthrene dication. However, the increased relative intensities of the bands associated with the minor isomer(s) allow the comparison with the theoretically predicted spectrum of 1102+ (Figure d). As can be seen, the hole-burning spectrum in the C–H stretching vibrations region becomes broader both on the red and the blue sides and shows a good agreement with the theoretical spectrum of 1102+.
The initially generated phenanthrene dication must undergo a complex rearrangement path to form the most stable isomer 1102+. The fact that the annealing to the lowest lying isomer prevails over the formation of other energetically preferred structures, such at 122+ (requiring one rearrangement) or anthracene dication (182 - requiring one rearrangement from 122+), suggests that the ionisation by high-energy electrons leads to dications at high energy state that can explore the whole potential energy surface and anneal to the most stable structure. This also could imply that the PAHs ions in the interstellar medium could be annealed via the collisions with high-energy electrons to the most stable structures.
We have further investigated the effect of anharmonicity on the calculated IR spectra of the PAH dications. Harmonic calculations failed to account for all experimentally observed bands in anthracene mono- and dication [Citation42]. Similarly, in the case of phenanthrene dication, the harmonic spectrum in Figure b does not predict several bands, namely the ones at 1333 and 1343.5 cm−1. Therefore, we performed anharmonic calculations for several candidates: 112+,1102+,122+,312+, 132+, and 1112+ (Figure ).
Figure 6. Calculated anharmonic spectra of a) phenanthrene dication 112+ and selected lowest energy structures, namely b) 1102+, c) 122+, d) 312+, e) 132+,and f) 1112+. The intensity of the theoretical bands in a) and b) mid-IR region are scaled for better visibility.
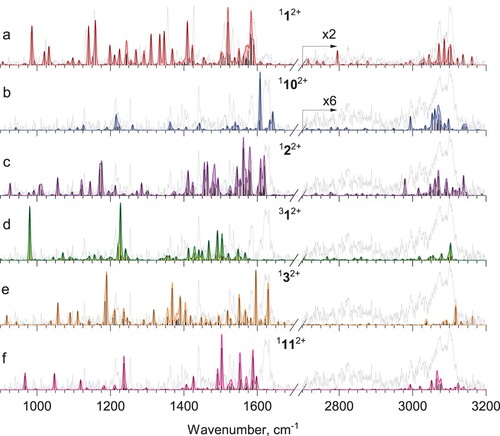
The anharmonic spectrum of a dication 112+ in Figure a matches very well with the experimentally observed bands in the spectrum of He-Phen2+. It is also evident that the spectrum of the most stable isomer, 1102+ matches most of the bands in the hole-burning spectrum albeit the intense bands at 1620 cm−1 ad 1585 cm−1 are shifted and predicted less accurately than by harmonic calculations (Figure b). The anharmonic IR spectra of the other structures do not fit with the experiment.
We thus conclude that the 1102+ dication is formed from phenanthrene dication during the ionisation with high-energy electrons. The most characteristic vibrational band of this isomer, 1625 cm−1 (6.2 µm), coincides with one of the observed aromatic infrared bands [Citation22–24], providing another evidence that this ion might be of high astrophysical interest.
We were unable to record the visible spectrum of the 1102 + isomer. Such an experiment requires a modified hole-burning design, where the Vis laser would be used as a probe, while the IR OPO/OPA would be used as a pump. This is challenging because of the low relative abundance of the second isomer in an initial ion ensemble and the low power of the IR OPO/OPA in the far-infrared region, prohibiting it from signal saturation. Another option is a Vis-Vis hole-burning tagging photodissociation spectroscopy that requires some instrumental modifications and will be conducted in the future.
Conclusions
We report a spectroscopic and theoretical study of small doubly-charged PAH phenanthrene. Using tagging photodissociation spectroscopy, we recorded the visible spectrum of this dication and showed that it is broad and unstructured, hence not a potential candidate for DIB search. The recorded IR spectrum revealed the presence of a minor isomer formed during the ionisation process with higher-energy electrons compared to previous studies. We explored several possible isomers, calculated their vibrational spectra, and compared them to the isomer-specific spectrum of a minor phenanthrene dication. Both harmonic and anharmonic calculations clearly indicate that the minor isomer corresponds to the most stable localised dication 1102+. Formation of this isomer requires substantial skeletal rearrangements of the initially formed phenanthrene dication. The absence of other low-energy lying isomeric dications suggests that the rearrangements proceed in high-internal energy states enabling complex rearrangements and lead to the annealing towards the most stable isomer on the potential energy surface. The fascinating features of the observed rearrangement are that the1102+ ion does not have a neutral counterpart and can only be formed in harsh conditions, such as the ISM, and its most intense vibrational band coincides with the one of the AIBs, making it highly appealing for further investigation.
Acknowledgement
This paper is dedicated to Dieter Gerlich, a great physicist and friend. The work was supported by the Netherlands Organisation for Scientific Research (NWO, 740.018.022).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- M.L. Heger, Lick Obs. Bull. 10, 146–147 (1922).
- D. Gerlich and S. Horning, Chem. Rev. 92, 1509–1539 (1992). doi:10.1021/cr00015a003
- D. Gerlich, J. Chinese Chem. Soc. 65, 637–653 (2018). doi:10.1002/jccs.201800122
- A. Pereverzev and J. Roithová, J. Mass. Spectrom. 57, e4826 (2022).
- M.A. Duncan, Int. J. Mass. Spectrom. 200, 545–569 (2000). doi:10.1016/S1387-3806(00)00366-3
- J. Oomens, B.G. Sartakov, G. Meijer and G. von Helden, Int. J. Mass. Spectrom. 254, 1–19 (2006). doi:10.1016/j.ijms.2006.05.009
- T.R. Rizzo, J.A. Stearns and O.V. Boyarkin, Int. Rev. Phys. Chem. 28, 481–515 (2009). doi:10.1080/01442350903069931
- T. Baer and R.C. Dunbar, J. Am. Soc. Mass. Spectrom. 21, 681–693 (2010). doi:10.1016/j.jasms.2010.01.028
- C.N. Stedwell, J.F. Galindo, A.E. Roitberg and N.C. Polfer, Annu. Rev. Anal. Chem. 6, 267–285 (2013). doi:10.1146/annurev-anchem-062012-092700
- N. Heine and K.R. Asmis, Int. Rev. Phys. Chem. 34, 1–34 (2015). doi:10.1080/0144235X.2014.979659
- L. Jašíková and J. Roithová, Chem. - A Eur. J. 24, 3374–3390 (2018). doi:10.1002/chem.201705692
- P.J. Sarre, J. Mol. Spectrosc. 238, 1–10 (2006). doi:10.1016/j.jms.2006.03.009
- E.B. Jochnowitz and J.P. Maier, Annu. Rev. Phys. Chem. 59, 519–544 (2008). doi:10.1146/annurev.physchem.59.032607.093558
- D. Gerlich, J. Jašík, D.V. Strelnikov and J. Roithová, Astrophys. J. 864, 62 (2018). doi:10.3847/1538-4357/aad630
- D.V. Strelnikov, J. Jašík, D. Gerlich, M. Murata, Y. Murata, K. Komatsu and J. Roithová, J. Phys. Chem. A. 122, 8162–8166 (2018). doi:10.1021/acs.jpca.8b06222
- D. Gerlich, J. Jašík and J. Roithová, Int. J. Mass. Spectrom. 438, 78–86 (2019). doi:10.1016/j.ijms.2018.12.018
- H. Linnartz, J. Cami, M. Cordiner, N.L.J. Cox, P. Ehrenfreund, B. Foing, M. Gatchell and P. Scheier, J. Mol. Spectrosc. 367, 111243 (2020). doi:10.1016/j.jms.2019.111243
- S. Leach, J. Electron. Spectros. Relat. Phenomena. 41, 427–438 (1986). doi:10.1016/0368-2048(86)85020-4
- K. Sellgren, Astrophys. J. 277, 623–633 (1984). doi:10.1086/161733
- K. Sellgren, L.J. Allamandola, J.D. Bregman, M.W. Werner and D.H. Wooden, Astrophys. J. 299, 416–423 (1985). doi:10.1086/163710
- M. Cohen, L. Allamandola, A.G.G.M. Tielens, J. Bregman, J.P. Simpson and F.C. Witteborn, Astrophys. J. 302, 737–749 (1986). doi:10.1086/164035
- A. Leger and J.L. Puget, Astrophys. 137, L5–L8 (1984).
- L.J. Allamandola, A.G.G.M. Tielens and J.R. Barker, Astrophys. J. 71, 733–775 (1989). doi:10.1086/191396
- D.M. Hudgins, L.J. Allamandola and S.A. Sandford, Adv. Sp. Res. 19, 999–1008 (1997). doi:10.1016/S0273-1177(97)00341-4
- H. Piest, G. von Helden and G. Meijer, Astrophys. J. 520, L75–L78 (1999). doi:10.1086/312143
- J.A. Piest, J. Oomens, J. Bakker, G. Von Helden and G. Meijer, Spectrochim. Acta - Part A Mol. Biomol. Spectrosc. 57, 717–735 (2001). doi:10.1016/S1386-1425(00)00439-X
- A.M. Ricks, G.E. Douberly and M.A. Duncan, Astrophys. J. 702, 301–306 (2009). doi:10.1088/0004-637X/702/1/301
- J. Oomens, A.J.A. van Roij, G. Meijer and G. von Helden, Astrophys. J. 542, 404–410 (2000). doi:10.1086/309545
- P. Bréchignac, T. Pino and N. Boudin, Spectrochim. Acta - Part A Mol. Biomol. Spectrosc. 57, 745–756 (2001). doi:10.1016/S1386-1425(00)00441-8
- G. Malloci, G. Mulas and C. Joblin, Astron. Astrophys. 426, 105–117 (2004). doi:10.1051/0004-6361:20040541
- I. Garkusha, J. Fulara, A. Nagy and J.P. Maier, Astrophys. J. 728, 131 (2011).
- M. Meyer, P. Martini, A. Schiller, F. Zappa, S.A. Krasnokutski and P. Scheier, Astrophys. J. 913, 136 (2021). doi:10.3847/1538-4357/abf93e
- J. Zhen, A. Candian, P. Castellanos, J. Bouwman, H. Linnartz and A.G.G.M. Tielens, Astrophys. J. 854, 27 (2018). doi:10.3847/1538-4357/aaa7f2
- J. Bouwman, C. Boersma, M. Bulak, J. Kamer, P. Castellanos, A.G.G.M. Tielens and H. Linnartz, Astron. Astrophys. 636, 1–9 (2020). doi:10.1051/0004-6361/201937013
- J. Zhen, P. Castellanos, J. Bouwman, H. Linnartz and A.G.G.M. Tielens, Astrophys. J. 836, 28 (2017). doi:10.3847/1538-4357/836/1/28
- U. Jacovella, C. Rossi, C. Romanzin, C. Alcaraz and R. Thissen, Chem. Phys. Chem 202200474, 1–8 (2022).
- J. Roithová, A. Gray, E. Andris, J. Jašík and D. Gerlich, Acc. Chem. Res. 49, 223–230 (2016). doi:10.1021/acs.accounts.5b00489
- J. Jašík, D. Gerlich and J. Roithová, J. Phys. Chem. A. 119, 2532–2542 (2015). doi:10.1021/jp5088064
- N. Godbout, D.R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem. 70, 560–571 (1992). doi:10.1139/v92-079
- F. Blanco, M. Solimannejad, I. Alkorta and J. Elguero, Na and Mg. Theor. Chem. Acc. 121, 181–186 (2008). doi:10.1007/s00214-008-0462-3
- M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M. Robb, J.R. Cheeseman, G. Scalmani, V.P.G.A. Barone, G.A. Petersson, H.J.R.A. Nakatsuji and X. Li, Gaussian 16, Revision A. 03, Gaussian. Inc., Wallingford CT, 3. 2016.
- J. Roithová, J. Jašík, J.J. Del Pozo Mellado and D. Gerlich, Faraday Discuss. 217, 98–113 (2019). doi:10.1039/C8FD00196K
- S. Banhatti, J. Palotás, P. Jusko, B. Redlich, J. Oomens, S. Schlemmer and S. Brünken, Astron. Astrophys. 648, 1–12 (2021). doi:10.1051/0004-6361/202039744
- O. Dopfer, Int. Rev. Phys. Chem. 22, 437–495 (2003). doi:10.1080/0144235031000112878
- J. Jašík, J. Žabka, J. Roithová and D. Gerlich, Int. J. Mass Spectrom. 354–355, 204–210 (2013).
- R. Navrátil, J. Jašík and J. Roithová, J. Mol. Spectrosc. 332, 52–58 (2017). doi:10.1016/j.jms.2016.10.016
- J. Jašík, D. Gerlich and J. Roithová, J. Am. Chem. Soc. 136, 2960–2962 (2014). doi:10.1021/ja412109h
- H.T. Jonkman and W.C. Nieuwpoort, Tetrahedron Lett. 14, 1671–1674 (1973). doi:10.1016/S0040-4039(01)96024-1
- K. Krogh-jespersen, J. Am. Chem. Soc, 417–423 (1991). doi:10.1021/ja00002a006