
Abstract
Background. Topical formulations of nonsteroidal anti-inflammatory drugs (NSAIDs) are generally considered to be safer alternatives to oral NSAIDs due to lower systemic absorption. We conducted randomized, crossover studies that compared the pharmacokinetics (PK), bioequivalence and safety of topical diclofenac sodium 2% twice daily (BID), diclofenac sodium 1.5% four times daily (QID) and oral diclofenac sodium in healthy subjects. Methods. The results of three bioequivalence studies are reviewed. Healthy adult subjects (n = 76) applied topical diclofenac sodium 2% solution (40.4 mg/2 mL) BID; or 1.5% solution (19.3 mg/40 drops) QID to each knee for 7.5 consecutive days separated by a washout period. Subjects (n = 22) in one study also received oral diclofenac sodium 75 mg BID for 7.5 days. Plasma diclofenac concentrations were determined from serial blood samples collected on Days 1 and 8 (steady state), and diclofenac PK parameters were estimated by noncompartmental methods. Results. The studies demonstrated comparable bioequivalence between the 2% and 1.5% topical solutions as well as lower systemic exposure compared to oral dosing (approximately 93% less). Daily systemic exposure was comparable between the two formulations with only a 12% difference in the AUCss0-24 (p = 0.140). Furthermore, both topical solutions demonstrated delayed elimination with a t1/2 of 4- to 6-fold longer, as compared to oral diclofenac. The 2% solution provided more consistent dosing relative to the 1.5% solution when comparing AUCss0-24 and Cmaxss across studies. Mild application site reactions were the most common treatment-emergent adverse event reported with topical diclofenac. Conclusions. The steady-state PK profile of topical diclofenac 2% solution administered BID is similar to that of the 1.5% solution administered QID. Systemic exposure to diclofenac is substantially lower after topical application as compared to oral administration. (Study 2 was registered with ClinicalTrials.gov; NCT01202799; https://clinicaltrials.gov/ct2/results?term=01202799&Search=Search).
Introduction
Osteoarthritis (OA), the most common form of arthritis, can cause pain, swelling and reduced joint mobility and is associated with impaired health-related quality of life [Citation1,2]. OA can affect any joint, but usually occurs in the hands, knees, hips or spine. Nonsteroidal anti-inflammatory drugs (NSAIDs) are frequently used for the treatment of OA because of their anti-inflammatory and analgesic activity, which is attributed, in part, to inhibition of prostaglandin biosynthesis [Citation3]. However, orally administered NSAIDs are associated with serious and potentially fatal gastrointestinal and cardiovascular complications. Risks of these complications are increased in patients with comorbidities, the elderly, and in patients taking certain other medications including, but not limited to, antithrombotic agents, corticosteroids, and selective serotonin reuptake inhibitors [Citation4,5]. In particular, elderly patients are at risk of serious toxicity from systemically administered NSAIDs. As a result, it is recommended that oral NSAIDs should either be avoided [Citation6], or used at the lowest effective dose for the shortest period of time possible [Citation7,8].
Topical formulations of NSAIDs such as diclofenac are a good alternative to oral NSAIDs in patients with knee OA [Citation8] because systemic exposure is greatly reduced and the effects on platelets are negligible after topical application compared with oral administration [Citation9]. Consequently, the adverse event (AE) profile is favorably altered when NSAIDs are applied topically. For example, when diclofenac is applied as a 1.5% topical solution, it is as effective as orally administered diclofenac at relieving the symptoms of knee OA [Citation10]; however, a pooled analysis of data from 927 patients enrolled in two 12-week clinical trials showed that treatment with topical diclofenac sodium 1.5% was associated with a significantly lower incidence of gastrointestinal AEs than treatment with oral diclofenac in patients with knee OA [Citation11]. Topical treatment was also associated with a lower incidence of cardiovascular AEs, serious AEs (SAEs) and laboratory abnormalities in this analysis [Citation11].
A new topical formulation of diclofenac sodium, i.e. diclofenac sodium 2% topical solution, has been developed with the objective of reducing the required frequency of applications from four times daily (QID) to twice daily (BID) and the hope of more consistent dosing and exposure. The composition of the new 2% topical formulation of diclofenac sodium is similar to that of the 1.5% solution; however, the higher concentration allows for delivery of a similar daily dose with fewer applications. In addition, the inclusion of a thickener (hydroxypropyl cellulose) allows for ease of application with a metered-dose pump delivery device, and a higher ethanol concentration reduces the drying time so that the product remains where it is applied and is absorbed from the site of application.
The bioequivalence, pharmacokinetics (PK) and safety of the new 2% topical formulation of diclofenac sodium administered BID have been compared with that of the older 1.5% formulation administered QID in three phase 1, randomized, open-label, multiple-dose crossover studies in healthy volunteers. In one study, subjects also received oral diclofenac BID to allow for comparisons of the PK profile of diclofenac after oral and topical administration. Reported herein are the results of these three studies.
Materials and methods
Participants
Eligible subjects were healthy male or non-pregnant, non-lactating female volunteers aged 18 to 55 years, with a body mass index (BMI) of 19 to ≤ 30 kg/m2 (Studies 1 and 3) or 19 to ≤ 29 kg/m2 (Study 2) and normal electrocardiogram (ECG) findings. Female subjects were required to have had a negative pregnancy test and to be using effective contraception before and during the trial, or to be either surgically sterile or postmenopausal for ≥ 12 months prior to screening. Eligible subjects were able to provide written informed consent and were willing and able to comply with the requirements of the study protocol.
Subjects hypersensitive to diclofenac and those with uncontrolled cardiac, renal, hepatic, pulmonary or other systemic disease; documented gastrointestinal bleeding (except hemorrhoidal) within the preceding 6 months; abnormal hepatic, renal or hematologic laboratory test values; and/or a history of major knee surgery (at any time), or minor knee surgery within the previous year were excluded. Concurrent NSAID use was prohibited.
Study design and treatment
The PK, bioequivalence and safety of two topical diclofenac formulations were evaluated in three phase 1, single-center, randomized, open-label, multiple-dose, two- or three-period studies (). In each of the three studies subjects applied diclofenac sodium 2% topical solution (PENNSAID®, diclofenac sodium topical solution 2%, Horizon Pharma USA, Inc., Deerfield, IL, USA) (40.4 mg [2 mL] to each knee BID; total daily dose, 162 mg/8 mL) for 7.5 consecutive days during one period and diclofenac sodium 1.5% topical solution (PENNSAID®, diclofenac sodium topical solution 1.5%, Horizon Pharma USA, Inc., Deerfield, IL, USA) (19.3 mg [40 drops, ≈ 1.2 mL] to each knee QID; total daily dose, 154 mg/9.6 mL) for 7.5 consecutive days in another period. In one study (Study 2), subjects also received a diclofenac sodium 75 mg delayed release tablet (Sandoz Pharmaceuticals Inc., Princeton, NJ, USA) orally BID (total daily dose, 150 mg) for 7.5 days in one period.
Table 1. Study designs.
There was a minimum washout period of 15 (Study 1) or 21 (Studies 2 and 3) days between the last dose of one treatment and the first dose of another treatment in each of the three studies. The sequence of individual treatments was determined by a computer-generated randomization code.
Subjects received each dose of trial medication under direct observation with the date and time of application recorded. The topical diclofenac preparations were applied to a clean, dry knee. A single dose of the 2% formulation was dispensed as two 1 mL pumps, while a single dose of the 1.5% solution was dispensed as 40 drops. The solutions were dispensed first into the hand and then were spread evenly around the front, back, and sides of the one knee and then the other, after which the skin was allowed to dry completely. Medication containers were weighed before and after each dose application to determine the amount of diclofenac delivered.
Blood samples and pharmacokinetic assessments
Blood samples (6 mL) for diclofenac plasma concentration analysis were collected predose (within 90 minutes prior to dosing) and then at 1, 2, 3, 4, 6, 8 and 12 hours post-dose on Day 1, before the first dose of medication (within 90 minutes prior to dosing) on Days 2 through 7, then at 0.5, 1, 2, 3, 4, 6, 6.5, 7, 8, 9, 10 and 12 hours post-dose on Day 8, and then once daily on Days 9, 13 and 15.
The time and date of collection for each sample were recorded. Blood samples were immediately placed into an ice bath and then centrifuged at approximately 4°C. The plasma fraction was divided equally into two labeled polypropylene tubes and frozen at or below –20°C within 1 hour of collection and remained frozen during shipping and until assayed.
Plasma samples were analyzed by PPD Bioanalytical Laboratories (Richmond, VA, USA) using a validated liquid chromatography/tandem mass spectrometry (LC-MS/MS) assay for diclofenac. The method was validated over a calibration range of 0.100 to 100 ng/mL diclofenac using 0.250 mL of heparinized plasma. Human plasma containing diclofenac and the internal standard (diclofenac-d4) were acidified and extracted with an organic solvent. The organic layer was removed, evaporated to dryness, and then reconstituted prior to injection onto a high-performance liquid chromatography column.
Quantitation was performed using separate weighted (1/x2) linear least squares regression analyses generated from calibration standards. Calculations were performed using Analyst (Version 1.4.2) and PPD Assist LMS (Version 5).
Diclofenac PK parameters for each individual for each treatment were estimated by noncompartmental methods using Phoenix WinNonlin Version 6.2 (Certara, L.P., Princeton, NJ, USA). Plasma concentrations below the limit of quantification (BLQ) that occurred before the time of observed maximum plasma concentration (Tmax) were set to 0 unless they occurred between measureable serum concentrations, in which case they were set to missing. Plasma concentration values that were BLQ after Tmax were set to missing.
Diclofenac PK parameters calculated on Day 1 and/or at steady state (ss; Day 8) were maximum observed plasma concentration (Cmax); minimum observed plasma concentration (Cmin); average plasma concentration (Cavg); time to Cmax (Tmax); degree of fluctuation (DFL; [Cmaxss – CminSS]/Cavgss) and swing ([Cmaxss – Cminss]/Cminss); area under the plasma concentration-time curve (AUC) adjusted to 0 to 12 hours (AUC0–12) in Study 1 or 0 to 24 hours (AUC0–24) in Studies 2 and 3. In addition, the elimination rate constant (Kel) and terminal elimination half-life (t1/2) were calculated using data collected over Days 8 to 15.
Safety and tolerability assessments
Safety and tolerability were assessed by physical examinations, vital signs, ECGs, laboratory tests, spontaneous reports of AEs and skin irritation assessments (during periods when subjects received topical diclofenac preparations). Skin irritation was assessed on an ordinal scale ranging from 0 to 4 where 0 = no visible reaction or equivocal response; 0.5 = dryness or flaking; 1 = erythema; 2 = erythema with induration; 3 = erythema with induration and vesiculation with blisters ≤ 5 mm in diameter; and 4 = erythema with induration and vesiculation with blisters >5 mm in diameter. Subjects with skin irritation Grade >2 were to be withdrawn from the studies.
Ethics
The protocols conformed to the Declaration of Helsinki and were approved by an institutional review board operating in accordance with the principles and requirements described in the US Code of Federal Regulations. All subjects provided written informed consent prior to undergoing any study procedures. Analysis of the samples followed the principles of Good Laboratory Practice and PPD’s standard operating procedures. Study 2 was registered with ClinicalTrials.gov: NCT 01202799.
Statistical analysis
The sample size was based on the results of a prior randomized crossover bioavailability study in which 25 subjects received diclofenac 1.5% topical solution (PENNSAID®) and diclofenac 3% topical gel (Solaraze®). The results demonstrated that the bioavailability of diclofenac 1.5% solution was significantly lower than that of diclofenac 3% gel, as was to be expected. On the basis of these results, the target enrollment in Studies 1, 2 and 3 was 32, 30 and 32 subjects, respectively. Two populations were used in the analyses: 1) all dosed subjects for the safety analyses, and 2) subjects completing all dosing periods, i.e. completers, for the PK analyses.
In one study (Study 1) the extent of exposure to diclofenac (AUC) was calculated over a 12-hour period (AUC0–12), while exposure was calculated over a 24-hour period (AUC0–24) in the two remaining studies (Studies 2 and 3).
PK parameters were calculated using standard noncompartmental methods. Summary statistics were calculated for all PK parameters by treatment. The SAS mixed linear model procedure was used to perform analysis of variance (ANOVA) on each PK parameter, with sequence, treatment, period, and subjects nested within sequences as sources of variation. Days 1–7 and steady-state PK parameters (Cmax, AUC, Days 2–7 Cmin, Cminss, Cmaxss, Cavgss and AUCss0–12) were analyzed on a logarithmic scale using ANOVA, while Tmax, Tmaxss, Kel and t1/2 were analyzed without transformation. SAS general linear model methodology was used with sequence, drug, period, and subjects nested within sequences as sources of variation.
Least squares mean (LSM) differences between treatments and associated 90% confidence intervals (CIs) were calculated to yield estimated ratios of PK parameters for treatment comparisons. Estimated differences of geometric LSM values and 90% CIs in the log scale were exponentiated to yield estimated ratios of PK parameters for treatment comparisons in the original scale and 90% CIs of the ratios. The two treatments (diclofenac 2% and 1.5% topical solutions) were considered to be bioequivalent if the 90% CI of the geometric LSM ratio was fully contained within the range of 80–125%.
The Wilcoxon signed-rank test was used to compare untransformed PK parameters (Tmax, Tmaxss) and skin irritation scores.
Statistically significant differences were defined as p < 0.05.
Results
Studies 1, 2, and 3 enrolled 32, 30, and 32 subjects, respectively, of whom 30, 22 and 29 individuals completed treatment. The baseline characteristics of all treated patients and completers in each trial are presented in . Across the three studies, the mean age ranged from 34 to 41 years and BMI ranged from 25 to 26 kg/m2. Two subjects withdrew prematurely from Study 1 (one for an AE and one for other reasons), eight subjects withdrew prematurely from Study 2 (four for protocol violations, two for AEs and two for other reasons) and three subjects withdrew from Study 3 because of AEs (). In addition, data from five subjects were excluded from the PK analysis in Study 1 because predose diclofenac concentrations were >5% of Cmax; thus, 25 subjects were included in the PK analysis in this trial.
Table 2. Subject demographics.
Figure 1. Flow diagram of subject disposition: Panel (A) Study 1, Panel (B) Study 2, and Panel (C) Study 3. Note that five subjects were excluded from the pharmacokinetic (PK) analysis in Study 1 because predose diclofenac concentrations were >5% of maximum observed plasma concentration; thus, 25 subjects were included in the PK analysis.
Pharmacokinetics
Study 1
As shown in , BID application of diclofenac sodium 2% topical solution (40 mg/knee) produced somewhat higher systemic exposure to diclofenac (Cmax and AUC0–12) than QID application of the 1.5% formulation (19.3 mg/knee) both on Day 1 and at steady state (Day 8: Cmaxss and AUCss0–12) (p < 0.05). Mean AUC0–12 increased from 77.27 ng · h/mL to 204.58 ng · h/mL between Day 1 and Day 8 after application of the 2% solution and from 27.46 ng · h/mL to 141.49 ng · h/mL after application of the 1.5% solution. Mean Cmax increased from 12.16 ng/mL on Day 1 to 25.24 ng/mL at steady state (Day 8) after application of the 2% solution and from 2.3 ng/mL on Day 1 to 17.04 ng/mL at steady state after application of the 1.5% solution. By Day 2, the Cmin values for the 2% topical solution were not significantly different from those seen on Day 8, suggesting that steady-state levels are achieved very rapidly. The Cmin values for the 1.5% topical solution indicate that steady-state levels are achieved by Day 3. Cmin values and apparent plasma t1/2 were not significantly different between the two solutions.
Table 3. Summary of diclofenac pharmacokinetic parameters on Day 1 and Day 8 of dosing with topical diclofenac 2% solution (40 mg/knee) BID and topical diclofenac 1.5% solution (19.3 mg/knee) QID in Study 1. Only data from study completers are shown.
A comparison of the LSM AUC0–12 on Days 1 and 8 shows that the extent of exposure to diclofenac was higher after application of the 2% as compared to the 1.5% solution (), although the degree of intrasubject variability was quite high. DFL and swing were higher after BID application of the 2% topical solution as compared with QID application of the 1.5% topical solution, and both Cavgss and Cminss were higher after application of the former formulation (). The high inter-individual variability (2% solution = 54% and 1.5% solution = 65%) and intra-individual variability (52%) observed in this study are typical for topical formulations.
The dose applied to both knees ranged from 2.8 to 4.0 g for diclofenac 2% (target = 4 g, mean 3.87 ± 0.18 g) and from 1.7 to 3.5 g for diclofenac 1.5% (target = 2.6 g, mean 2.45 ± 0.31 g). No applications of diclofenac 2% exceeded the target dose.
Considering the high inter-individual variability typically observed (coefficient of variability [CV] >35%) with topical formulations, the two formulations can be considered to have comparable PK properties on Day 8. Any differences are not likely to be clinically relevant, due to the similar trough plasma concentrations (Cminss) for diclofenac 2% and 1.5% (9.94 and 8.83 ng/mL, respectively; p = 0.51) and given that the log-transformed geometric LSM values for DFL were comparable (88.48 and 69.76), with a CV of 21%.
Studies 2 and 3
In Studies 2 and 3, the peak (Cmax) and extent (AUC0–24) of systemic exposure to diclofenac was higher on Day 1 after BID application of the 2% solution than after QID application of the 1.5% solution (). Moreover, peak exposure (Cmax) was nearly identical on Day 1 after application of diclofenac 2% in Study 2 (15.63 ng/mL) and Study 3 (15.53 ng/mL), as was the extent of exposure (AUC0–24; 190.82 and 199.07 ng · h/mL, respectively; ).
Table 4. Summary of diclofenac pharmacokinetic parameters on Day 1 and Day 8 of dosing with topical diclofenac 2% solution (40.4 mg/knee) BID and topical diclofenac 1.5% solution (19.3 mg/knee) QID in Studies 2 and 3. Only data from study completers are shown.
In contrast to the results obtained on Day 1, systemic exposure at steady state (Cmaxss and AUCss0–24) was comparable for the two formulations on Day 8 ().
Diclofenac 2% topical solution demonstrated comparable systemic exposure at steady state (Day 8; ) with AUCss0–24 values of 315 and 323 ng · h/mL in Studies 2 and 3, respectively, as compared with diclofenac 1.5% (354 and 323 ng · h/mL, respectively). Likewise, Cmaxss values of approximately 20 ng/mL were obtained with diclofenac 2% in both studies and were comparable to those obtained with diclofenac 1.5% in Studies 2 (18 ng/mL) and 3 (15 ng/mL).
LSM AUC0–24 was significantly higher after application of the 2% solution on Day 1 in both Studies 2 and 3 (p < 0.0001). The difference in LSM AUCss0–24 between the two formulations was smaller on Day 8 and was not statistically significant in Study 2 (p = 0.6824 in Study 2 and 0.0352 in Study 3; ).
Systemic exposure estimates (AUCss0–24 and Cmaxss) on Day 1 and Day 8 in the two studies were more consistent (i.e. the variability was lower) for the 2% solution delivered by a metered-dose pump than the 1.5% solution, which is delivered as drops. Indeed, the estimated mean (standard deviation) AUCss0–24 and Cmaxss values for diclofenac were nearly identical for the 2% solution in the two studies (), which prompted us to pool the data.
The pooled analysis demonstrates that the 2% formulation produced a consistently higher diclofenac plasma concentration profile than the 1.5% formulation on Day 1, while the steady-state diclofenac plasma concentration profiles for the two formulations were similar (Day 8; ).
Figure 2. Mean concentration of diclofenac in plasma on Day 1 (A) and Day 8 (B) after topical application of diclofenac sodium 2% and 1.5% solutions. Pooled analysis of data from subjects who completed treatment in two trials.
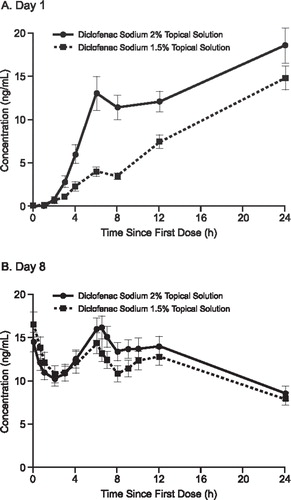
The pooled analysis also demonstrates that exposure to diclofenac was comparable after application of the 2% and 1.5% solutions when measured as Cmaxss (19.79 ± 10.11 ng/mL vs 16.10 ± 9.21 ng/mL) and as AUCss0–24 (319.51 ± 162.36 ng · h/mL vs 295.47 ± 168.91 ng · h/mL) (). As seen in , there were no statistically significant differences between the 2% and 1.5% solutions in any of the pooled outcomes at steady state (Day 8). The LSM difference between the two formulations for AUCss0–24 was 12% in the pooled analysis (p = 0.140).
Table 5. Summary of diclofenac pharmacokinetic parameters on Day 1 and Day 8 of dosing with topical diclofenac 2% solution (40.4 mg/knee) BID and topical diclofenac 1.5% solution (19.3 mg/knee) QID. Combined results of data from Studies 2 and 3. Only data from study completers are shown.
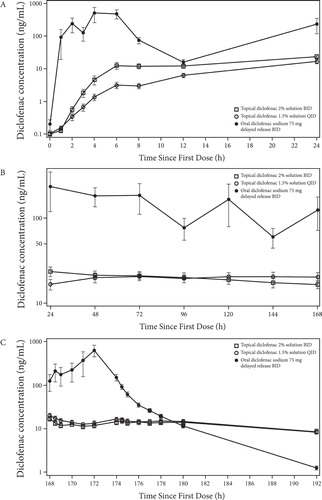
Exposure to diclofenac was substantially different after oral administration than after application of either topical formulation in Study 2 (; -). For example, at steady state, peak exposure (mean Cmaxss) was 67- to 84-fold lower after topical application than after oral administration of diclofenac (18−20 ng/mL vs 1348 ng/mL, respectively; ). Similarly, the extent of exposure (mean AUCss0–24) was 14- to 15-fold lower after topical as compared to oral administration (315−354 ng · h/mL vs 4426 ng · h/mL; ). Moreover, elimination was 4- to 6-fold slower after topical application than oral administration, as indicated by the marked difference in mean t1/2 between the two routes of administration in Study 2 (35−36 hours after topical application vs 6 hours after oral administration; ). These data indicate that the systemic exposure of the 2% solution is about 7% of that found with a comparable dose of oral diclofenac at steady state.
Figure 3. Mean plasma concentration profile of diclofenac after topical application of diclofenac sodium 2% and 1.5% solutions and oral diclofenac 75 mg in Study 2 from 0 to 24 hours (Day 1, A), 24 to 168 hours (Days 2 to 7, B) and 168 to 192 hours (Day 8, C).
Safety and tolerability
In general, application site reactions were the most common treatment-emergent AE (TEAE) reported during application of topical diclofenac (). Among application site reactions, dryness was the most common TEAE (reported by 22.7% to 46.9% of subjects after application of diclofenac 2% solution BID and 20.0% to 35.5% of subjects after application of diclofenac 1.5% solution QID; ).
Table 6. Safety and tolerability of topical diclofenac 2% solution applied BID and topical diclofenac 1.5% solution applied QID for 7.5 days in three studies
There were no statistically significant differences between mean skin irritation scores for the 2% and 1.5% solutions in any study. The maximum skin irritation scores reported by any subject after application of either topical formulation were 1, 0.5 and 3 in Studies 1, 2 and 3, respectively. Only one subject reported a skin irritation score >1 in any of the studies. A 38-year-old woman experienced erythema of both knees during treatment with diclofenac 2% in Study 3. Treatment was withdrawn as specified in the protocol and the subject received treatment with hydrocortisone cream (the skin irritation score was 1 for each knee the next day). Two other subjects withdrew from Study 3 due to an AE: one due to increased blood pressure; one due to excoriation. Constipation was reported as a TEAE after application of topical diclofenac in two of the three studies ().
No SAEs were reported in any of the three studies. A total of six subjects withdrew from the three studies because of AEs: one subject withdrew from Study 1 due to flu-like symptoms (unrelated to treatment); two subjects withdrew from Study 2, one because of a positive pregnancy test during treatment with diclofenac 2% (unrelated to treatment), and a second because of a rash during treatment with diclofenac tablets (related to treatment); three subjects withdrew from Study 3, one because of increased blood pressure during treatment with diclofenac 1.5% (possibly related to treatment), a second because of a right knee abrasion after a fall during treatment with diclofenac 2% (unrelated to treatment), and a third because of application site dermatitis during treatment with diclofenac 2% (related to treatment).
Discussion
The collective results of the three PK studies show that systemic exposure to diclofenac observed after BID application of the 2% solution is comparable to that observed after QID application of the 1.5% solution and is substantially lower than that observed after a comparable dose of BID administration of oral diclofenac. In each of the three studies, both the peak (Cmax) and extent (AUC) of systemic exposure to diclofenac on Day 1 were considerably higher after BID application of the 2% formulation as compared with QID application of the 1.5% formulation. In contrast, the plasma exposure profiles were more similar at steady state, and the pooled analysis of data from Studies 2 and 3 shows that there was no statistically significant difference in AUCss0–24 between the two formulations. As expected, both DFL and swing were higher after application of the 2% solution than after the 1.5% solution due to less frequent application. We reported more variability in the AUCs with the 1.5% solution as compared with the 2% solution when viewed across studies.
In Studies 2 and 3, the estimates of Cmax and AUC after application of the 2% solution were nearly identical on both Day 1 and Day 8, while there was more variability in the corresponding estimates of diclofenac exposure after application of the 1.5% solution. As the total daily dose of diclofenac was similar after application of the 2% and 1.5% solutions (161.6 mg vs 154.4 mg, respectively), the observed differences in exposure do not reflect differences in the amount of drug applied, but likely represent both the delivery technique and the BID administration as compared to the QID administration required by the 1.5% solution. The formulations are similar and contain the same amount of dimethyl sulfoxide, a powerful penetrating agent which facilitates absorption through the skin. The 2% solution has a slightly higher alcohol content (drying agent) and contains hydroxypropyl cellulose which increases the viscosity of the solution relative to the 1.5% solution. These attributes may encourage more consistent absorption with the 2% solution over time. Nonetheless, our results indicate that overall exposure is similar between the two formulations.
We found that systemic exposure to diclofenac was substantially decreased after topical, as compared with oral, administration in Study 2. Under steady-state conditions, peak exposure (Cmax) was as much as 84-fold lower and the extent of exposure (AUCss0–24) was approximately 15-fold lower after application of either topical formulation than after oral administration of a comparable dose (150 mg/day) with a commercial dosage form. In addition to these marked differences in exposure, the t1/2 of diclofenac was substantially longer after application of either topical formulation than after administration of the oral dosage form (approximately 35 hours vs approximately 6 hours). These differences in exposure and t1/2 suggest that, in contrast to oral administration, diclofenac is not rapidly and extensively absorbed into the systemic circulation after topical application, but rather a depot is established at the site of application from which the drug is slowly released. The rapid stabilization of diclofenac concentrations and delayed elimination after topical application may minimize the effect of dosing inconsistencies.
The BID dosing regimen for diclofenac 2% topical solution should result in improved adherence with the treatment regimen because of increased convenience, which may in turn enhance the effectiveness of treatment. This supposition is based on analyses that have demonstrated significantly better adherence when oral medications are administered BID as compared with more frequent administration [Citation12,13].
The 2% solution also has characteristics that may improve convenience. Unlike the 1.5% solution, it contains hydroxypropyl cellulose, which increases the viscosity of the preparation, and has a higher concentration of ethanol, which results in more rapid drying after application. Collectively, these properties prevent running and dripping, and may result in better compliance, retention, and penetration of diclofenac at the site of application.
The dose delivery technique differs between the two formulations. The 2% solution is administered with a metered-dose pump that delivers 1 mL per actuation; thus, two actuations are sufficient to deliver the dose required to treat one knee. In contrast, the 1.5% solution is applied as 40 drops per knee, which, when combined with the more frequent application regimen, is less convenient for patients or caregivers. The pump used to deliver the 2% formulation may also provide a more accurate and consistent dose than the container for the 1.5% solution. In the studies reported in this paper, the variability in dosing was minimized by having trained study personnel deliver doses into the hands of volunteers. In routine clinical practice, the higher variability in exposure to diclofenac after application of the 1.5% solution may be exacerbated by counting errors or functional limitations.
The greatly reduced systemic exposure after topical application is reflected in a tolerability profile that is markedly different from that associated with oral administration [Citation10]. In patients with OA, oral administration of diclofenac is associated with a higher incidence of gastrointestinal and cardiovascular AEs, SAEs and laboratory abnormalities when compared with topical application of the 1.5% formulation. In contrast, topical application of the 1.5% formulation is associated primarily with local AEs [Citation11]. Nonetheless, all NSAIDs, including nonselective and COX-2 selective agents as well as topical formulations such as those evaluated in these studies, may increase the risk of serious cardiovascular events, stroke, and gastrointestinal bleeding [Citation14]. Thus, NSAIDs should be used with caution in patients with existing cardiovascular disease or cardiovascular risk factors and in those with a history of gastrointestinal bleeding.
The three studies included in this report were conducted in healthy volunteers; however, the results suggest that the tolerability profile of the new 2% formulation is similar to that of the older 1.5% formulation. The safety and efficacy of the older formulation has been compared with that of oral diclofenac in randomized trials in patients with OA. Collectively, the results of these clinical trials show that topical diclofenac has similar efficacy when compared with oral diclofenac, but is associated with a much lower incidence of systemic AEs (including gastrointestinal, cardiovascular and hepatic AEs) and SAEs [Citation11]. As a result, the use of topical diclofenac rather than oral diclofenac should result in significant reductions in morbidity and mortality associated with NSAID treatment in patients with OA.
Both diclofenac topical preparations were well tolerated by subjects across the three studies. Dryness at the site of application was the most common AE in each study and mean skin irritation scores were consistently ≤ 1 throughout each study. Only one individual had a skin irritation score of 3 (for erythema), which mandated withdrawal from treatment. The erythematous reaction resolved rapidly after withdrawal of treatment and application of topical hydrocortisone.
Conclusion
In conclusion, the observed PK profile for diclofenac sodium 2% topical solution administered BID in three studies was similar to that of the 1.5% formulation administered QID. Both topical formulations share the advantage of substantially lower systemic exposure to diclofenac and substantially longer t1/2 than orally administered diclofenac. The safety and tolerability of the two formulations were similar. Thus, the collective results suggest that diclofenac sodium 2% topical solution provides a similar PK profile with greater convenience than the 1.5% topical solution and can be safely and effectively administered BID to patients with OA of the knee.
Declaration of interest
Blair Jarvis and Alan J. Klopp, PhD, of inScience Communications, Springer Healthcare, provided medical writing support funded by Horizon Pharma. This manuscript was supported by Horizon Pharma. This study was supported by Horizon Pharma and Mallinckrodt Pharmaceuticals. RJ Holt has received consulting fees from Horizon Pharma USA, Inc, Novartis, POZEN Pharmaceuticals, Inc, Cadence Pharmaceuticals, Inc, Hospira, and is a previous employee of Searle/Pfizer. He has no financial interest in any of these companies. JD Kent and T Taiwo are current employees of Horizon Pharma USA, Inc. and are holders of stock in Horizon Pharmaceuticals. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Notes
References
- Zhang W, Doherty M, Peat G, Bierma-Zeinstra MA, Arden NK, Bresnihan B, et al. EULAR evidence-based recommendations for the diagnosis of knee osteoarthritis. Ann Rheum Dis 2010;69:483–9
- Silverwood V, Blagojevic-Bucknall M, Jinks C, Jordan JL, Protheroe J, Jordan KP. Current evidence on risk factors for knee osteoarthritis in older adults: a systematic review and meta-analysis. Osteoarthritis Cartilage 2015;23:507–15
- Vane JR, Botting RM. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am J Med 1998;104:2S–8S; discussion 21S-2S
- Crooks CJ, West J, Card TR. Comorbidities affect risk of nonvariceal upper gastrointestinal bleeding. Gastroenterology 2013;144:1384–93; quiz e18-9.
- Masclee GM, Valkhoff VE, Coloma PM, de Ridder M, Romio S, Schuemie MJ, et al. Risk of upper gastrointestinal bleeding from different drug combinations. Gastroenterology 2014;147:784–92; quiz e13-4
- American Geriatrics Society Panel on Pharmacological Management of Persistent Pain in Older Persons. Pharmacological management of persistent pain in older persons. J Am Geriatr Soc 2009;57:1331–46
- Altman RD, Barthel HR. Topical therapies for osteoarthritis. Drugs 2011;71:1259–79
- Zhang W, Moskowitz RW, Nuki G, Abramson S, Altman RD, Arden N, et al. OARSI recommendations for the management of hip and knee osteoarthritis, Part II: OARSI evidence-based, expert consensus guidelines. Osteoarthritis Cartilage 2008;16:137–62
- Kienzler JL, Gold M, Nollevaux F. Systemic bioavailability of topical diclofenac sodium gel 1% versus oral diclofenac sodium in healthy volunteers. J Clin Pharmacol 2010;50:50–61
- Simon LS, Grierson LM, Naseer Z, Bookman AA, Zev Shainhouse J. Efficacy and safety of topical diclofenac containing dimethyl sulfoxide (DMSO) compared with those of topical placebo, DMSO vehicle and oral diclofenac for knee osteoarthritis. Pain 2009;143:238–45
- Roth SH, Fuller P. Diclofenac topical solution compared with oral diclofenac: a pooled safety analysis. J Pain Res 2011;4:159–67
- Claxton AJ, Cramer J, Pierce C. A systematic review of the associations between dose regimens and medication compliance. Clin Ther 2001;23:1296–310
- Richter A, Anton SE, Koch P, Dennett SL. The impact of reducing dose frequency on health outcomes. Clin Ther 2003;25:2307–35; discussion 6
- PENNSAID (diclofenac sodium topical solution 2%). Highlights of presribing information. Horizon Pharma USA, Inc; Deerfield, IL: 2015