
Abstract
α-thalassemia is one of the most common monogenic diseases worldwide and is caused by reduced or absent synthesis of α-globin chains, most commonly due to deletions of one or more of the α-globin genes. α-thalassemia occurs with high frequency in tropical and subtropical regions of the world and are very rarely found in the indigenous Scandinavian population. Here, we describe four rare forms of α-thalassemia out of which three are novel, found in together 20 patients of Norwegian origin. The study patients were diagnosed during routine hemoglobinopathy evaluation carried out at the Department of Medical Biochemistry, Oslo University Hospital, Norway. The patients were selected for their thalassemic phenotype, despite Norway as country of origin. All samples went through standard hemoglobinopathy evaluation. DNA sequencing and copy number variation (CNV) analysis using quantitative real-time polymerase chain reaction (qPCR) was applied to detect sequence variants and uncommon deletions in the α-globin gene cluster, respectively. Deletion breakpoints were characterized using gap-PCR and DNA sequencing. DNA sequencing revealed a single nucleotide deletion in exon 3 of the HBA2 gene (NM_000517.4(HBA2):c.345del) and a novel deletion of 20 nucleotides in exon 2 of the HBA2 gene (NM_000517.4(HBA2):c.142_161del). qPCR CNV analysis detected two novel large deletions in the α-globin gene cluster, –(NOR) deletion covering both α-globin genes and (αα)Aurora Borealis affecting the regulatory region, leaving the downstream α-globin genes intact. Even though inherited globin gene disorders are extremely rare in indigenous Scandinavians, the possibility of a carrier state should not be ignored.
Introduction
α-thalassemia is one of the most common human genetic diseases worldwide and is caused by reduced or absent production of α-globin chains [Citation1]. The duplicated α-globin genes, HBA2 and HBA1, are located at the telomeric region at chromosome 16 (16p13.3), and the expression is strictly regulated by a region located ∼25–65 kilobase pairs (kb) upstream of the genes [Citation2]. This region consists of four multispecies conserved sequences (MCSs), called MCS R1- R4. MCS R2, also called HS-40, has been shown to be the human α-globin major regulatory element and essential for expression of the α-globin genes [Citation2,Citation3]. The molecular basis of α-thalassemia is usually large deletions in the α-globin gene cluster involving one (α+) or both (α0) α-globin genes [Citation4]. Deletions involving the regulatory region HS-40, small insertions and deletions, and sequence variants that inactivate one of the linked α-globin genes are less frequent. However, an increasing number of sequence variants and small deletions and insertions resulting in reduced production of α-globin chains are reported. Due to natural selection through relative resistance of heterozygous individuals against severe malaria, inherited hemoglobin (Hb) disorders have a high gene frequency in many tropical and sub-tropical countries [Citation5]. α-thalassemia occurs at a high frequency in Mediterranean countries, South-East Asia, Africa, the Middle East, and the Indian subcontinent [Citation1]. As a consequence of migration during the past decades, α-thalassemia and other inherited Hb disorders have been introduced in most parts of Northern and Western Europe [Citation1]. However, for people of Scandinavian origin thalassemia are extremely rare and may easily be confused with iron deficiency. Multiplex gap polymerase chain reaction (gap-PCR) is widely used to detect common deletions causing α-thalassemia [Citation6–8]. However, gap-PCR is restricted to those deletions where the breakpoints are known; hence, the method is not suitable for the detection of unknown deletions. Several applications have been developed to detect rare and unknown deletions in the α-globin gene cluster. Most used is multiplex ligation-dependent probe amplification, and numerous rare deletions have been identified using this approach [Citation9–13]. Recently, we developed a copy number variation (CNV) application based on quantitative real-time PCR (qPCR) [Citation14], which was introduced in our second-line diagnostics of hemoglobinopathies applied for those cases suspected of having α-thalassemia despite normal multiplex α-thalassemia gap-PCR (α-thal gap-PCR). Here, we describe an amended form of the qPCR-CNV application (HBA-CNV) containing four additional hydrolysis probes (TaqMan® CNV assays). Furthermore, four rare forms of α-thalassemia in 20 patients from eleven different families of Norwegian origin, detected with qPCR CNV analysis or DNA sequencing, are presented. Three of these mutations are described for the first time.
Materials and methods
Patients and routine analyses
The study patients were diagnosed during routine hemoglobinopathy evaluation carried out between 2012 and 2018 at the Department of Medical Biochemistry, Oslo University Hospital, Norway. The patients were selected for their thalassemic phenotype, despite Norway as country of origin. The Regional Committees for Medical and Health Research Ethics approved the study (ethical agreement REK 2015/2352) and written informed consent was obtained from all the included subjects.
Ethylenediaminetetraacetic acid (EDTA) whole blood from the patients was sent to our laboratory for hemoglobinopathy evaluation. All samples went through first-line diagnostics, including (i) complete blood count (CBC) (2012–2014): Sysmex XE-2100, Sysmex, Cobe, Japan, 2014–2018: Sysmex XN-9000), (ii) Hb fractionation performed by high-performance liquid chromatography analysis (2012–2014: beta-thalassemia short program, Variant, Bio-Rad, Hercules, USA, 2014–2018: Variant II, BioRad) and/or capillary electrophoresis (Capillarys 2 Flex Piercing, Sebia, Evry Cedex, France), (iii) measurement of plasma ferritin and C-reactive protein concentrations (Cobas 8000, e602, Roche Diagnostics, Risch-Rotkreuz, Switzerland), and (iv) multiplex α-thal gap-PCR detecting the seven most common deletions causing α-thalassemia, described elsewhere [Citation8]. For some of the samples, standard evaluation (CBC, Hb fractionation, and α-thal gap-PCR) were performed at other medical laboratories in Norway with inconclusive results, before our evaluation. Next, the samples were submitted to second-line diagnostics including qPCR CNV analysis and/or DNA sequencing of the α-globin genes.
DNA extraction
Genomic DNA (gDNA) was prepared from 200 µL EDTA whole blood using the MagNA Pure LC DNA Isolation Kit I protocol according to the manufacturer’s instruction (Roche Applied Science, Penzberg, Germany), and eluted in a final volume of 100 µL. gDNA concentrations were determined using Nano Drop ND-1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA). gDNA was diluted to a final concentration of 30 ng/µL before gap-PCR and DNA sequencing and to 3.0 ng/uL before qPCR CNV analysis.
DNA sequencing of the globin genes
Amplification of the α-globin genes, HBA2 (NM_000517.4) and HBA1 (NM_000558.4), were performed on a Thermal Cycler (VWR, Radnor, PA) using HotStar Taq DNA Polymerase Kit (Qiagen, Venlo, Netherlands) and dNTP mix PCR Grade (Qiagen) according to the manufacturer’s instructions. In short, PCR was performed using 30 ng gDNA, 0.2 µM forward and reverse primer, 200 µM of each dNTP, 1× Q-solution, 1× PCR Buffer, and 2.5 U/reaction of HotStarTaq DNA Polymerase in a final volume of 50 µL. The cycling conditions comprised 15 min of enzyme activation at 95 °C followed by 35 cycles of 94 °C for 1 min, 60 °C for 1 min and 72 °C for 1 min 30 s, and a final extension step at 72 °C for 10 min. PCR products were purified and sequenced in both directions using an ABI 3730 high-throughput capillary electrophoresis instrument (Thermo Fisher Scientific) at the University of Oslo (from 2012 to 2014) and the Department of Medical Genetics at Oslo University Hospital (from 2014 to 2018). PCR and sequencing primers are shown in . Sequence data were analyzed using the program FinchTV v.1.4.0 (Geospiza Inc, Seattle, WA, USA) and HbVar tracks in the UCSC Genome Browser [Citation15,Citation16] (2012–2017) and SeqScape Software Version 2.7 (Thermo Fisher Scientific) and Alamut®Visual version 2.4 (Interactive Biosoftware, Rouen, France) (2017–2018).
Table 1. Primer sequences for DNA sequencing, break point analysis, gap-PCR, and CNV analysis.
α-Thalassemia qPCR CNV analysis (HBA-CNV)
To detect CNVs in the α-globin gene cluster, a qPCR CNV analysis developed by our group was performed from 2013 to 2015 as described by Grimholt et al. [Citation14]. To improve the quality and reliability of the interpretation, two pre-designed TaqMan® CNV assays (Hs03937725_cn and Hs00290821_cn, Thermo Fisher Scientific) and two custom made TaqMan® CNV assays () were added to amplify in proximity to each of the four existing assays described by Grimholt et al. [Citation14]. Custom-made CNV assays were designed using Primer Express Software, version 1 (Thermo Fisher Scientific), and delivered by Thermo Fisher Scientific. The samples were analyzed in a final volume of 10 µL using 4 µL diluted gDNA, 5 µL TaqMan® Universal PCR MasterMix (Thermo Fisher Scientific), 0.5 µL TaqMan® Copy Number Reference Assay, RNase P (Thermo Fisher Scientific), and 0.5 µL TaqMan® Copy Number Assay in a 384 wells plate using Viia7 Real-Time PCR Instrument (Thermo Fisher Scientific). The amplification curves were analyzed as described elsewhere [Citation14].
Breakpoint analysis
To estimate the minimum length of the two novel deletions, two additional panels (panel X and Y, ) with pre-designed CNV TaqMan® assays (Thermo Fisher Scientific) located on both sides of the predicted deletions were analyzed using the same conditions as described above. The exact breakpoints of the –(NOR) deletion were characterized by gap-PCR with primers located on each side of the deletion. Based on the results of the qPCR CNV analysis, several unique primers were designed in the area between the position of the last CNV assay showing two copies and the first CNV assay showing one copy (indicating deletion). Primers that successfully generated a PCR product covering the breakpoint are listed in (Primer BP-F and BP-R). Primers were designed using Primer 3 v.0.4.0 [Citation17,Citation18] and PCR was performed using LongRange PCR Kit (Qiagen) according to the manufacturer’s instructions. The cycling conditions comprised 3 min polymerase activation at 93 °C followed by 35 cycles of 93 °C for 15 s, 56 °C for 30 s and 68 °C for 3 min. PCR products were separated and analyzed on a GelStar® (Lonza, Basel, Switzerland) stained 2% agarose gel. Direct sequencing of the PCR product containing the breakpoint fragment was performed as described for DNA sequencing of the globin genes.
Table 2. TaqMan® assays were used for CNV analysis for the α-globin gene cluster and flanking regions.
–(NOR) gap-PCR
When the breakpoints of the –(NOR) deletion were identified, a specific gap-PCR was developed to confirm the –(NOR) deletion in patients of Norwegian origin where the HBA-CNV panel showed one copy of the A3–A8 assays (). Primers flanking the deletion were designed to generate an amplicon of 545 bp in the presence of the –(NOR) deletion (Primer NOR-F and NOR-R, ). PCR was performed using HotStar Taq DNA Polymerase Kit (Qiagen) and dNTP mix PCR Grade (Qiagen) as described for DNA sequencing of the globin genes. The cycling conditions comprised 15 min of enzyme activation at 95 °C followed by 35 cycles of 94 °C for 1 min, 60 °C for 1 min and 72 °C for 1 min, and a final extension step at 72 °C for 10 min. PCR products were separated and analyzed on GelStar® (Lonza) stained 2% agarose gel.
Results
Samples from patients suspected of having α-thalassemia despite their Norwegian origin were analyzed using DNA sequencing or qPCR CNV analysis of the α-globin gene cluster in addition to first-line diagnostics as described in Materials and Methods. Common to all patients in the study were persistent microcytosis and normal iron status (). Hb fractionation showed a normal Hb pattern in all cases, excluding high Hb A2 β-thalassemia and common Hb variants. None of the seven common α-thalassemia deletions tested for by the α-thal gap-PCR were detected in any of the patients. DNA sequencing revealed one previously described single nucleotide (nt) deletion in exon 3 of HBA2 (HBA2:c.345del), and a novel 20 bp deletion in exon 2 of HBA2 (HBA2:c.142_161delGACCTGAGCCACGGCTCTGC). qPCR CNV analysis displayed two novel large deletions, one of them covering both α-globin genes (–(NOR)), and deletion of the upstream regulatory region, including HS-40 ((αα)Aurora Borealis).
Table 3. Hematological, biochemical, and molecular data of the patients studied.
HBA2:c.345delC
The single nt deletion HBA2:c.345delC was detected in seven patients from six different families. None of these patients were anemic, but all showed microcytosis and elevated or high-normal red blood cell (RBC) count (). In patients showing hematological changes consistent with α+-thalassemia, i.e. mean corpuscular Hb (MCH) above 23 pg and Hb concentration within reference limits [Citation1] and that obtain negative α-thal gap-PCR, the cascade reflex testing normally starts with DNA sequencing of the α-globin genes. The index case presented with erythrocytosis, MCH 24 pg and Hb 133 g/L (P1-1, ). DNA sequencing revealed a heterozygous deletion of a single nt in exon 3 of HBA2, causing a frameshift and a premature termination signal at codon 132 (globin nomenclature) (). The deletion included one of four identical nucleotides (cytosine) and based on the performed analysis it was not possible to decide which of the four cytosines were deleted. According to the Human Genome Variation Society (HGVS) guidelines [Citation19], the deletion was reported based on the so-called 3′ rule, assigning it to the most 3′ nt of the stretch. In addition to the HGVS nomenclature, we used the conventional numbering systems for the globin genes [Citation15]. Thus, the sequence variant was designated NM_000517.4(HBA2):345del, p.(Ala116Profs*18) (HGVS nomenclature) or HBA2:c.345delC, alpha2 114(-C); modified C-terminal sequence: (114) Pro-Pro-Ser-Ser-Pro-Leu-Arg-Cys-Thr-Pro-Pro-Trp-Thr-Ser-Ser-Trp-Leu-Leu-(132) COOH (globin nomenclature), and was entered into the HbVar database [Citation15] (HbVar ID 3055). The single nt deletion was previously reported as HBA2:c.342delC in the IthaGenes database (IthaID 403) [Citation20]. Moreover, the sequence variant was registered as rs746988006 in dbSNP [Citation21] and reported in ClinVar [Citation22] as pathogenic.
Figure 1. (A) Schematic representation of the small deletions found in HBA2 (NM_000517.4). Solid boxes denote the exons, solid black lines introns and hatched boxes represent the 5′ and the 3′ untranslated regions. The arrows show the location of the two small deletions found in exon 2 and exon 3 of HBA2. (B) Sequencing of the HBA2 gene showing a deletion of a single nt in exon 3 causing a frameshift and a PTC in codon 132. (C) A 20 nt deletion in exon 2 produces a new reading frame starting at codon 47 and ends in a PTC three positions downstream.
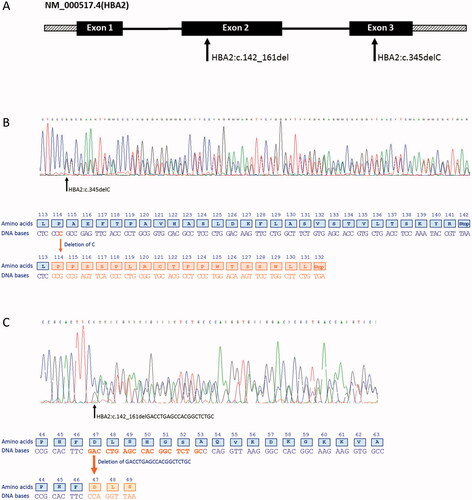
HBA2:c.142_161delGACCTGAGCCACGGCTCTGC
In one patient, sequencing of the α-globin genes revealed a heterozygous deletion of 20 nucleotides (GACCTGAGCCACGGCTCTGC) in exon 2 of HBA2 (). No abnormalities in the HBA1 gene were found. First-line diagnostics showed microcytosis and erythrocytosis with a normal Hb pattern and no sign of iron deficiency (). The α-thal gap-PCR was negative. The 20 nt deletion created a frameshift starting at codon 47 (globin nomenclature [Citation15]) and the new reading frame ended in a premature termination codon (PTC) three positions downstream (). The deletion was designated NM_000517.4(HBA2):142_161del, p.(Asp48Profs*3) (HGVS nomenclature) or HBA2:c.142_161delGACCTGAGCCACGGCTCTGC (in short: HBA2:c.142_161del). The variant was entered into the HbVar database (HbVar ID 3180) [Citation15].
–(NOR) deletion
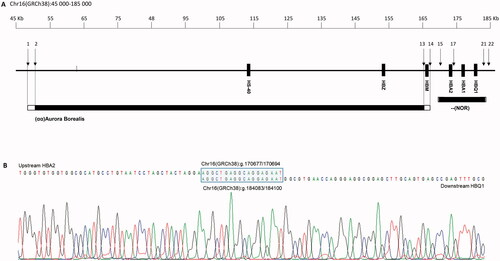
The novel α0-deletion () was found in eight patients from three different families, all of Norwegian origin. All patients showed microcytosis with MCH ranging from 21 to 25 pg (). The index case was a 58-year-old woman who presented with persistent microcytosis. She had received iron treatment for a long time without effect. Despite the patient’s ethnicity, the general practitioner referred the patient to hemoglobinopathy evaluation. First-line diagnostics showed a normal Hb pattern, MCH 22 pg, slight anemia, and RBC in the upper part of the reference interval (Patient VII-I, ), indicative of an α0-thalassemia [Citation1]. Based on the results, the cascade reflex testing started with qPCR CNV analysis [Citation14], which revealed the deletion of both α-globin genes. To further characterize the deletion, a selection of pre-designed CNV TaqMan® assays (Panel X, ) were used to narrow the breakpoints to a 3.3 kb region upstream of HBA2 and a 1.2 kb region downstream of HBQ1. Breakpoint analysis with several unique primers was applied to identify the exact breakpoint. The primer pair, BP-F, and BP-R () successfully produced an amplicon of ∼2 kb. Sequencing of the amplicon pinpointed the breakpoint sequence to an 18 nt homologous sequence (AGGCTGAGGCAGGAGAAT), making this a 13.4 kb deletion that begins upstream of HBA2 (position Chr16(GRCh38):g.170677/170694) and ends downstream of HBQ1 (position Chr16(GRCh38):g.184083/184100) (). The deletion was entered into the HbVar database as –(NOR) (HbVar ID 3021). Later, the –(NOR) deletion was identified in several family members of the index patient and patients from two other Norwegian families. In these patients, the specific –(NOR) gap-PCR was applied to confirm the deletion after HBA-CNV analysis (data not shown).
Figure 2. (A) Schematic representation of part of 16p13.3 showing a 190 kb region containing the α-globin gene cluster and flanking regions. The solid boxes denote the α-globin like genes and the regulatory region (HS-40). Black bars show the two deletions, –(NOR) and (αα)Aurora Borealis. The open ends indicate the region where the breakpoint is located. Black arrows and numbers indicate the location of the probes. (B) Characterization of breakpoints by gap-PCR and direct sequencing of –(NOR) showed that the deletion starts upstream of HBA2 at position 170677/170694 and ends at position 184083/184100, downstream of HBQ1. The blue box is the 18 nt homologous sequence where the homologous recombination event occurs.
(αα)Aurora borealis
The (αα)Aurora Borealis deletion was first detected in a 39-year-old woman of Norwegian origin. First-line diagnostics performed at a different medical laboratory gave inconclusive results and the sample was sent to us for further analysis. The HBA-CNV panel revealed a heterozygous deletion removing the complete regulatory region, including HS-40, leaving the downstream α-globin genes intact (). Any additional molecular defects in the α-globin genes that might contribute to the phenotype were excluded by DNA sequencing (data not shown). Later, the deletion was detected in two other family members. All presented with a typical thalassemic phenotype with microcytosis and erythrocytosis (). A panel of TaqMan® assays (Thermo Fisher Scientific) distributed from NM_016310.4(POLR3K) to NM_001003938.3(HBM) (panel Y, ) showed that the deletion covered a region of minimum 115.2 kb (Chr16(GRCh38):g.∼50379 ∼ 165612). Unfortunately, gap-PCR analysis to pinpoint the exact breakpoints of the deletion was unsuccessful, hence the approximate size of the deletion is grounded on the most 5′ CNV assay showing one copy (Hs03941017_cn, located Chr16(GRCh38)50379) and the most 3′ CNV assay showing one copy (Hs03948127_cn, located Chr16(GRCh38)165612). The maximum length of the deletion, based on the most 5′- and 3′ CNV assays showing two copies, is 119.4 kb.
Discussion
The Department of Medical Biochemistry at Oslo University Hospital is a de facto reference laboratory of hemoglobinopathy evaluation in Norway and annually we receive more than 1200 samples from both primary and secondary health care all over the country. An ongoing study at our laboratory shows that ∼90% of the patients with α-thalassemia show positive α-thal gap-PCR [Citation23]. To be able to clarify the molecular cause of the remaining 10%, either DNA sequencing of the α-globin genes, HBA-CNV, or both are required. Together with the present study, this shows that qPCR CNV is a valuable supplementary in the routine diagnostics of hemoglobinopathies, along with DNA sequencing. The second line diagnostics works like cascade reflex testing. Depending on the findings in the first-line diagnostics, one or more reflexive tests may be required to provide a clinical interpretation. In most cases, the degree of microcytosis depends roughly on the number of α-globin genes affected and correlates well with the reduction in α-globin chain synthesis predicted for each mutant [Citation1]. As mean corpuscular volume (MCV) increases rapidly during storage [Citation24], the mean corpuscular hemoglobin (MCH) value is a more reliable parameter to evaluate the degree of microcytosis and works as a key diagnostic parameter in the cascade reflex testing. In general, HBA-CNV is requested in those cases suspected of having an α-thalassemia that remains uncharacterized after α-thal gap-PCR with an MCH below 23 pg. If the HBA-CNV is negative, DNA sequencing of the α-globin genes is requested. If the MCH value is above 23 pg, it is more reasonable to believe that the cause of the microcytosis is a sequence variant in one of the two α-globin genes, thus DNA sequencing is requested.
For targeted detection of disease genes and their regulatory regions, RT-qPCR is a perfectly suited method because of low costs, no post-PCR processing, and a fast turnaround time [Citation25,Citation26]. To increase the efficiency of the HBA-CNV method and make the interpretation of the results more reliable, a 384 wells format was applied and four additional hydrolysis probes were implemented to improve the previously described method [Citation14]. Because of the low resolution of the HBA-CNV method, a disadvantage is that double gene deletions with breakpoints located within or close to the 5- or 3-prime end of HBA2 or HBA1, respectively, such as the –(α)20.5 deletion, might incorrectly be interpreted as a single gene deletion [Citation27]. The possibility of a double gene deletion should always be considered when the HBA-CNV application shows one copy of the HBA3.7 assays alone, or together with one copy of the HBA2- or HBA1 assays. To avoid misinterpretations, a thorough evaluation of haematological data together with CNV data is pivotal.
Although inherited Hb disorders, including α-thalassemia, traditionally is endemic among populations originating from tropical or subtropical regions of the world, massive population movement over the few past decades has spread these disorders to all parts of the world [Citation1]. Norway has for the last 40 years experienced increasing immigration from Africa and Asia. There are now (2021) more than 400,000 people of Asian or African descent living in Norway [Citation28]. Hence, hemoglobinopathies are not a rarity in Norway anymore, but thalassemia found in patients originating from Norway is extremely rare. Nevertheless, in 1975, Jakobsen et al. [Citation29] described 12 patients in two Norwegian families with β-thalassemia minor, and in 2009 Brantberg et al. [Citation30] described a Norwegian family with severe neonatal anemia across two generations due to εγδβ-thalassemia. The first Norwegian patient with α-thalassemia was described in 2008 [Citation31], when Joly and coworkers detected a single nt deletion (–C) in codon 113 of the HBA2 gene, introducing a PTC in position 132. Transcripts containing PTC located more than 50–55 base pairs upstream of the last exon-exon boundary are selectively eliminated by nonsense-mediated decay (NMD) [Citation32]. This surveillance system affects the clinical phenotype by preventing the deleterious effect of some truncated proteins, e.g. unstable α-globin chains.
HBA2:c.345delC
A single nt deletion, HBA2:c.345delC, creating a termination signal position 132, the same position as for the frameshift variant described by Joly et al., was first described in a patient of unknown ethnicity through newborn screening in 2006 [Citation33]. Our study describes seven individuals in six different families of Norwegian origin heterozygous of HBA2:c.345del, all presented with microcytosis and mild erythrocytosis (). Although this may be indicative of thalassemia, several of them were initially thought rather have an iron deficiency because of their Norwegian ethnicity. Iron deficiency was later excluded and hemoglobinopathy evaluation could confirm a non-deletional α+-thalassemia. HBA2:c.345delC would most likely escape NMD and α-globin chains with a putative disrupted H helix would be translated. The G and H helices are critical for the α1β1 binding site and the binding of alpha hemoglobin stabilizing protein (AHSP) [Citation34,Citation35], and sequence variants located in these helices may lead to unstable α-globin chain variants with a thalassemic phenotype [Citation35,Citation36]. AHSP is a molecular chaperone that binds free α-globin chains and inhibits their precipitation within erythrocyte precursors in the bone marrow, preventing apoptosis and ineffective erythropoiesis [Citation34,Citation37]. The single nt deletion, HBA2:c.345delC, occurs in the GH corner of the α-globin chain and is predicted to disrupt the amino acid sequence in the H-helix and the translation ends in codon 132 (H15). Due to impaired binding to AHSP, the unstable α-globin chain will be degraded before Hb assembly, explaining the thalassemic phenotype of the patients. Several Hb variants have been described causing a thalassemic phenotype due to impaired binding to AHSP [Citation31,Citation33,Citation35,Citation36,Citation38]. The frameshift variant in Hb Lynwood (HBA2:c.323delT) [Citation38], results in a PTC in the same position as HBA2:c.345del. Hb Lynwood was found in conjunction with the -α 3.7 deletion in a male patient with hematological changes comparable with coinheritance of two α+-thalassemias (αNDα/-α, ND means non-deletional) (MCH 20 pg, RBC 6.00 × 1012 and Hb 124 g/L) [Citation1,Citation38].
HBA2:c.142_161delGACCTGAGCCACGGCTCTGC
A frameshift variant in exon two of HBA2 was found in a Norwegian patient with microcytosis and mild erythrocytosis. Frameshift variants may be assumed to disrupt gene function by leading to the complete absence of the gene product due to lack of transcription or NMD of an altered transcript [Citation39]. HBA2:c.142_161del creates a PTC more than 50–55 bp upstream of the last exon-exon boundary (codon 50, exon 2) and would probably undergo NMD, resulting in haploinsufficiency [Citation32]. Splice site prediction tools embedded in Alamut®Visual v2.4 suggest activation of a cryptic acceptor splice site in codon 49, adjacent to the PTC. To see whether this cryptic acceptor site is activated further analysis is needed. The change HBA2:c.142_161del represents the third small deletion described in codon 48 of HBA2, the previously described being HBA2:c.143del [Citation40] and HBA2:c.143_146del [Citation41], both causing a PTC and a and probably targets for NMD.
Using the American College of Medical Genetics and Genomics (ACMG) standards and guidelines for the interpretation of sequence variants [Citation39], both variants, HBA2:c.345delC and HBA2:c.142_161del, can be classified as pathogenic variants. They are both absent in the Norwegian population database ‘1000 Norwegian genomes’ [Citation42] as well as in a local patient database containing ∼500 exomes provided by the Department of Medical Genetics at Oslo University Hospital. Microcytosis and erythrocytosis in the absence of iron deficiency and β-thalassemia, are indicative of a carrier trait for α-thalassemia; hence the phenotype supports the notion that the variants are pathogenic. Moreover, null variants (e.g. frameshift) disrupt gene function and are considered as very strong evidence of pathogenicity [Citation39].
–(NOR) deletion
Large deletions are the most common cause of α-thalassemia. Although the single gene deletion -α3.7 is the most frequent deletion worldwide, a higher number of different double gene deletions than single-gene deletions have been identified. Ninety-five deletions causing α-thalassemia (ranging from 0.2 to 10.0 kb in size) are registered in the IthaGenes database [Citation20], 68 of them covering at least both α-globin genes. Five of the most common double gene deletions (–SEA, –MED, –THAI, –FIL, –(α)20.5) and two of the most common single-gene deletions (–α3.7, –α4.2) are easily detectable with gap-PCR [Citation8], but other deletions will remain undetected by this method. By using our HBA-CNV application based on qPCR and the ΔΔCq method, a previously unknown double gene deletion, designated –(NOR), was first found in three generations of a Norwegian family, all presented with a thalassemic phenotype. Double gene deletions are most frequently a result of non-homologous recombination [Citation43]. Nevertheless, several deletions caused by homologous recombination are described in the literature [Citation44–46]. Recombination between homeologous (partially homologous) Alu elements represent a major form of genetic instability in the human genome leading to deletions and duplications [Citation47]. Sequencing of the breakpoints of –(NOR) showed that both breakpoints are located within Alu elements and an 18 nt homologous sequence occurring at both the 5′ and 3′ regions (). This indicates that the deletion is a result of a simple crossover between homeologous Alu elements that are normally ̴13.4 kb apart.
(αα)Aurora borealis
The regulatory region of the α-globin like genes consists of several MCSs acting as binding sites for important erythroid transcription factors, such as GATA1, GATA2, NE-F2, and KLFs [Citation3]. Through a variety of expression studies and naturally occurring deletions in patients, the HS-40 (MCS-R2) has proven to be the most important region [Citation3]. The first deletion of the HS-40 region was described in a patient with α-thalassemia in 1990 by Hatton et al. [Citation48]. Since then, several deletions of the varying range have been described, 14 of them summarized by Higgs and Wood [Citation3]. The (αα)Aurora Borealis deletion described in our patients covers a region of a minimum of 115 kb, deleting all four MCSs.
The hematological changes observed in α-thalassemia may be confused with iron deficiency, especially in indigenous Northern Europeans, where inherited globin disorders are extremely rare. If a patient has persistent microcytosis and erythrocytosis with normal levels of ferritin and CRP, the possibility of a carrier state should not be ignored based on the patient’s ancestry. In our study, molecular analysis (sequencing, qPCR CNV, or both) was required in all samples to confirm the α-thalassemia diagnosis. Appropriate genetic testing is important to explain the hematological observations in the patients and to provide adequate genetic counselling for at-risk couples.
Acknowledgements
The authors would like to thank the biomedical laboratory scientists at the Department of Medical Biochemistry, Oslo University Hospital for their expert technical assistance. The authors would also like to thank the staff at the Unit for Cardiac and Cardiovascular Genetics, Department of Medical Genetics, Oslo University Hospital for their help with DNA sequencing.
Disclosure statement
The authors report no conflicts of interest. The authors are responsible for the content and writing of this article.
References
- Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010;5:13.
- Higgs DR. The molecular basis of α-thalassemia. Cold Spring Harb Perspect Med. 2013;3(1):a011718.
- Higgs DR, Wood WG. Long-range regulation of alpha globin gene expression during erythropoiesis. Curr Opin Hematol. 2008;15(3):176–183.
- Farashi S, Harteveld CL. Molecular basis of α-thalassemia. Blood Cells Mol Dis. 2018;70:43–53.
- Williams TN, Weatherall DJ. World distribution, population genetics, and health burden of the hemoglobinopathies. Cold Spring Harb Perspect Med. 2012;2(9):a011692.
- Chong SS, Boehm CD, Higgs DR, et al. Single-tube multiplex-PCR screen for common deletional determinants of alpha-thalassemia. Blood. 2000;95(1):360–362.
- Liu YT, Old JM, Miles K, et al. Rapid detection of alpha-thalassaemia deletions and alpha-globin gene triplication by multiplex polymerase chain reactions. Br J Haematol. 2000;108(2):295–299.
- Tan AS-C, Quah TC, Low PS, et al. A rapid and reliable 7-deletion multiplex polymerase chain reaction assay for alpha-thalassemia. Blood. 2001;98(1):250–251.
- Harteveld CL. State of the art and new developments in molecular diagnostics for hemoglobinopathies in multiethnic societies. Int J Lab Hematol. 2014;36(1):1–12.
- Traeger-Synodinos J, Harteveld CL. Advances in technologies for screening and diagnosis of hemoglobinopathies. Biomark Med. 2014;8(1):119–131.
- Colosimo A, Gatta V, Guida V, et al. Application of MLPA assay to characterize unsolved α-globin gene rearrangements. Blood Cells Mol Dis. 2011;46(2):139–144.
- Harteveld CL, Voskamp A, Phylipsen M, et al. Nine unknown rearrangements in 16p13.3 and 11p15.4 causing alpha- and beta-thalassaemia characterised by high resolution multiplex ligation-dependent probe amplification. J Med Genet. 2005;42(12):922–931.
- Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30(12):e57.
- Grimholt RM, Urdal P, Klingenberg O, et al. Rapid and reliable detection of α-globin copy number variations by quantitative real-time PCR. BMC Hematol. 2014;14(1):4.
- Patrinos GP, Giardine B, Riemer C, et al. Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies. Nucleic Acids Res. 2004;32(Database issue):D537–D541.
- Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res. 2002;12(6):996–1006.
- Untergasser A, Cutcutache I, Koressaar T, et al. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115.
- Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23(10):1289–1291.
- den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37(6):564–569.
- Kountouris P, Lederer CW, Fanis P, et al. IthaGenes: an interactive database for haemoglobin variations and epidemiology. PLOS One. 2014;9(7):e103020.
- Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–311.
- Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–d1067.
- Fjeld B, Sudmann-Day AA, Grimholt RM, et al. Alpha-thalassemia (manuscript in preparation). Oslo, Norway: Department of Medical Biochemistry, Oslo University Hospital; 2021.
- Sudmann-Day AA, Piehler A, Klingenberg O, et al. Six-day stability of erythrocyte and reticulocyte parameters in-vitro: a comparison of blood samples from healthy, iron-deficient, and thalassemic individuals. Scand J Clin Lab Invest. 2015;75(3):247–253.
- Weaver S, Dube S, Mir A, et al. Taking qPCR to a higher level: analysis of CNV reveals the power of high throughput qPCR to enhance quantitative resolution. Methods. 2010;50(4):271–276.
- D'Haene B, Vandesompele J, Hellemans J. Accurate and objective copy number profiling using real-time quantitative PCR. Methods. 2010;50(4):262–270.
- Grimholt RM, Urdal P, Klingenberg O, et al. Correction to: Rapid and reliable detection of α-globin copy number variations by quantitative real-time PCR. BMC Hematol. 2019;19:13.
- Statistisk sentralbyrå (Statistics Norway); 2021. Available from: http://www.ssb.no/befolkning/statistikker/innvbef
- Jakobsen E, Godal HC, Kierulf P. Thalassaemia minor. Twelve patients in two Norwegian families. Acta Med Scand. 1975;197(1–2):19–25.
- Brantberg A, Eik-Nes SH, Roberts N, et al. Severe intrauterine anemia: a new form of epsilongammagammadeltabeta thalassemia presenting in utero in a Norwegian family. Haematologica. 2009;94(8):1157–1159.
- Joly P, Pegourie B, Courby S, et al. Two new alpha-thalassemia point mutations that are undetectable by biochemical techniques. Hemoglobin. 2008;32(4):411–417.
- Khajavi M, Inoue K, Lupski JR. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur J Hum Genet. 2006;14(10):1074–1081.
- Eng B, Patterson M, Walker L, et al. Three new alpha-thalassemia point mutations ascertained through newborn screening. Hemoglobin. 2006;30(2):149–153.
- Feng L, Gell DA, Zhou S, et al. Molecular mechanism of AHSP-mediated stabilization of alpha-hemoglobin. Cell. 2004;119(5):629–640.
- Thom CS, Dickson CF, Gell DA, et al. Hemoglobin variants: biochemical properties and clinical correlates. Cold Spring Harb Perspect Med. 2013;3(3):a011858.
- Wajcman H, Traeger-Synodinos J, Papassotiriou I, et al. Unstable and thalassemic alpha chain hemoglobin variants: a cause of Hb H disease and thalassemia intermedia. Hemoglobin. 2008;32(4):327–349.
- Vasseur-Godbillon C, Marden MC, Giordano P, et al. Impaired binding of AHSP to alpha chain variants: Hb groene hart illustrates a mechanism leading to unstable hemoglobins with alpha thalassemic like syndrome. Blood Cells Mol Dis. 2006;37(3):173–179.
- Finlayson J, Ghassemifar R, Holmes P, et al. Hb lynwood [α107(G14) (-T) (α2) HBA2:c.323delT)] in conjunction with the α(3.7) deletion produces a moderately severe α-thalassemia phenotype. Hemoglobin. 2011;35(2):142–146.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424.
- Finlayson J, Ghassemifar R, Holmes P, et al. α-thalassemia trait caused by frameshift mutations in exon 2 of the α2-globin gene: HBA2:c.131delT and HBA2:c.143delA. Hemoglobin. 2012;36(5):511–515.
- Henderson SJ, Timbs AT, McCarthy J, et al. Ten years of routine α- and β-globin gene sequencing in UK hemoglobinopathy referrals reveals 60 novel mutations. Hemoglobin. 2016;40(2):75–84.
- The Norwegian 1000 genomes project; 2017 [cited 2017 Oct 12]. Available from: http://kreftgenomikk.no/en/1000genomes/
- Nicholls RD, Fischel-Ghodsian N, Higgs DR. Recombination at the human alpha-globin gene cluster: sequence features and topological constraints. Cell. 1987;49(3):369–378.
- Coelho A, Picanco I, Seuanes F, et al. Novel large deletions in the human alpha-globin gene cluster: clarifying the HS-40 long-range regulatory role in the native chromosome environment. Blood Cells Mol Dis. 2010;45(2):147–153.
- Harteveld KL, Losekoot M, Fodde R, et al. The involvement of Alu repeats in recombination events at the alpha-globin gene cluster: characterization of two alphazero-thalassaemia deletion breakpoints. Hum Genet. 1997;99(4):528–534.
- Jia SQ, Li J, Mo QH, et al. Alpha0 thalassaemia as a result of a novel 11.1 kb deletion eliminating both of the duplicated alpha globin genes. J Clin Pathol. 2004;57(2):164–167.
- White TB, Morales ME, Deininger PL. Alu elements and DNA double-strand break repair. Mob Genet Elements. 2015;5(6):81–85.
- Hatton CS, Wilkie AO, Drysdale HC, et al. Alpha-thalassemia caused by a large (62 kb) deletion upstream of the human alpha globin gene cluster. Blood. 1990;76(1):221–227.