
Abstract
Objectives: To assess the influence of smoking on histological disease severity and fibrosis in real-world NAFLD patients.
Material and methods: Consecutive NAFLD patients were identified with liver biopsies performed between 2008 and 2015. Characteristics such as smoking status and total number of pack years were collected. Biopsies were revised and BRUNT fibrosis and NAFLD activity score (NAS) determined. Patients with a high NAS (≥5) were compared to patients with a low NAS (<5) and with advanced fibrosis (stage 3–4) to patients with no-early fibrosis (stage 0–2). Patients with a history of smoking (current or past smoker) were defined ever smokers.
Results: Fifty-six patients were included (mean age 49 ± 14.3, 68.9% males and 39.3% history of smoking). Ever smokers had a higher fibrosis score than never smokers; two (IQR 0–3) versus one (IQR 1–1.5) (p = .040). Patients with advanced fibrosis smoked significantly more pack years than patients with no-early fibrosis; 10.6 (IQR 0–25.8) versus 0 (IQR 0–7) (p = .011). There is a weak to moderate correlation between fibrosis stage and number of pack years (Spearman’s Rho = 0.341, p = .012). There was no difference in NAS between never and ever smokers; 2.8 ± 1.5 versus 3.3 ± 1.4 (p = .205). Patients with NAS <5 had a median number of pack years of 0 (IQR 0–9) versus a median of 10.3 pack years (IQR 0–24) in patients with NAS ≥5 (p = .127).
Conclusion: Smoking is associated with severity of NAFLD-related liver fibrosis but not with histological disease severity. This supports the recommendation to cease smoking for NAFLD patients.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most expanding cause of chronic liver disease. NAFLD consists of a spectrum of conditions, ranging from simple steatosis to nonalcoholic steatohepatitis (NASH) and ultimately end-stage liver cirrhosis [Citation1]. The diagnosis of NASH is defined by the presence and pattern of specific histological abnormalities on liver biopsy, such as inflammation. For this, a system of scoring the individual features of NAFLD, the NAFLD Activity Score (NAS) was developed [Citation2]. This score is based on three histological parameters, namely lobular inflammation, macroscopic steatosis and ballooning of hepatocytes.
Advanced stages of NAFLD have been associated with risk factors such as diabetes, obesity and increasing age [Citation3–5]. Whether smoking aggrevates chronic liver diseases and especially NAFLD has been increasingly subject of research debate.
In obese rats, smoking increased the histological severity of NAFLD, favoring the development of NASH by aggrevation of lobular inflammation and hepatocellular ballooning [Citation6]. The underlying mechanisms seem to be induction of hepatocellular apoptosis, oxidative stress and modulation of several signaling pathways, such as ERK phosphorylation and AKT activation. The effect of smoking on fibrosis was less clear in this study. There was upregulation of several fibrogenic genes (pro-collagen α-2 and TIMP-1), but no significant fibrosis deposition in the animals.
In human NAFLD, smoking seems to be associated with severe fibrosis, as shown in a large cohort study encompassing 1091 patients, collected from enrollment in several studies [Citation7]. Long-term smoking was significantly associated with the presence of advanced fibrosis, suggesting cigarette smoking may accelerate disease progression. However, in this study, the association between smoking, NASH and the histological severity of independent NAS components was not extensively studied. In a brief report by Yilmaz et al, no association was observed between smoking and severity of NAFLD [Citation8].
Since human results are conflicting and experimental data suggest smoking can lead to progression from simple NAFLD to NASH, we conducted the present human cohort study. We aim to investigate whether there is an association between smoking and the presence of NASH, including the individual histological disease components; lobular inflammation, hepatocellular ballooning, steatosis and severity of fibrosis.
Methods
Patient population
All liver pathology reports from 2008 to 2009 in the VU University Medical Center and from 2010 to 2015 in the Radboud university medical center were screened for inclusion, if the words NAFLD, NASH or steatosis were mentioned in the report. Patient details were included when the diagnosis nonalcoholic fatty liver disease or nonalcoholic steatohepatitis was made on clinical and histological grounds. Patients were excluded when there was concomitant liver disease. Patient characteristics such as age, sex, length, weight, body mass index (BMI), the presence of diabetes mellitus type 2 (DM) and alcohol use (units/week) were assessed. This study was approved by the Institutional Review Boards of both hospitals.
Liver histology
All biopsies were stained with hematoxylin–eosin and assessed for fibrosis using Masson’s Trichome or Elastin-Von Gieson. In each center, the included biopsies were revised by a single liver pathologist, blinded for clinical information. Quality of biopsies was assessed and biopsies not meeting the following requirements were excluded: 20–25 mm long and/or containing more than 11 portal tracts. The stage of fibrosis was scored with the histological scoring system for NAFLD developed by Brunt et al [Citation2]. No or early fibrosis was defined as stage 1 or 2 and advanced fibrosis was defined as stages 3 or 4. The NAFLD Activity Score (NAS) was used to assess the severity of histological disease activity, with a total score between 0–8 based on individual scores for the three components; steatosis (0–3), lobular inflammation (0–3) and hepatocellular ballooning (0–2). A NAS of five or higher is defined as histological steatohepatitis [Citation9].
Smoking history
Smoking history was obtained from chart review and when unclear from direct interview with patients. Patients were informed with a study information letter and afterwards telephoned for an interview. From the data obtained from this interview and medical records, the total number of pack years smoked by each patient was calculated (one pack year equals one pack of cigarettes each day during one year). Patients were furthermore classified as ‘never smokers’ or ‘ever smokers’, the latter in case of current smoking or a history of smoking.
Statistical analysis
Continuous data are presented with mean and standard deviation (±SD), nominal data as counts (n), percentages and ordinal data with medians and interquartile ranges (IQR). Independent t-test or nonparametric tests where applicable were used to compare continuous variables. Chi-square test was used to compare categorical variables and Mann–Whitney U-test for ordinal variables. Correlations were tested with Pearson’s correlation coefficient or Spearman’s rho, depending on the level of measurement. A two-sided level of p<.05 was considered statistically significant. All statistical analyses were carried out using SPSS software (version 22; SPSS Inc., Chicago, IL).
Results
Patient characteristics
A total of 160 pathology reports with the words NAFLD, NASH or steatosis in the report were screened. Of these 76 were excluded because of a different or concomitant disease. Of 27 more patients, no sufficient smoking data were present. Finally, four biopsies were excluded because of bad quality. Leading to a total of 56 patients included in the analysis, with a mean age of 49 ± 14.3 years and 68.9% males. Of all patients, 22 (39.3%) had a history of smoking (named hereafter ever smokers). This group comprised of six subjects (27.3%) who still actively smoke and 16 subjects (72.7%) who ceased smoking. Mean number of pack years among ever smokers was 16.3 ± 10.8. Never smokers were significantly younger than ever smokers (p .028), but BMI, the presence of DM and alcohol use was not different between groups (, near here). There was no correlation between age and the number of pack years smoked by ever smokers (Pearson’s R = 0.169, p = .028).
Table 1. Patient characteristics.
Liver histology
The median fibrosis score according to Brunt was significantly higher in ever smokers than in never smokers; 2 (IQR 0-3) versus 1 (IQR 1–1.5) (p = .040). Whereas most never smokers had no or early fibrosis, subjects who ever smoked had predominantly early to advanced fibrosis. This was however not significant. There was no difference in the NAS between never and ever smokers (p = .205) ().
Number of pack years and severity of fibrosis
Overall, patients with advanced fibrosis smoked significantly more pack years than patients with no-early fibrosis; 10.6 (IQR 0–25.8) versus 0 (IQR 0–7) (p = .011) (). This observation was not altered after exclusion of never smokers: patients with advanced fibrosis smoked 22.3 ± 11 pack years, whereas patients with no-early fibrosis smoked 13 ± 9.4 pack years (p = .048) (). There is a weak to moderate correlation between the stage of fibrosis and the number of pack years smoked by all included patients (Spearman’s rho = 0.341, p = .012) and a similar trend for ever smokers (Spearman’s rho = 0.416, p = .061).
Figure 1. Number of pack years compared between patients with no-early (stage 0–2) and advanced fibrosis (stage 3–4). Panel A shows the difference in number of pack years (represented with boxplots with median, IQR and min–max value) for the whole cohort. Panel B shows the difference in number of pack years (represented as mean + standard error of the mean) for ever smokers (active and past smokers).
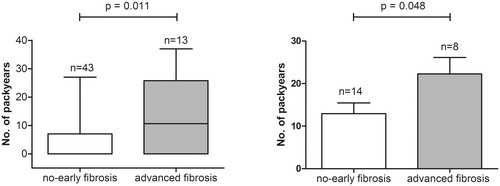
Possible confounders in advanced fibrosis
The mean NAS was higher in patients with advanced fibrosis compared to no-early fibrosis; 3.9 ± 1 versus 2.7 ± 1.5 (p = .003). In detail, a higher, but not significant, proportion of patients with advanced fibrosis had a NAS of 5 or higher compared to patients with no-early fibrosis (30.8% vs. 9.3%; p = .074). Patients with advanced fibrosis were older (mean age 60.4 ± 9.7 years vs. 45.5 ± 13.7 years; p < .001).
No other clinical features, such as sex, BMI, type-2 DM or alcohol use differed between patients with no-early fibrosis compared to advanced fibrosis (data not shown).
Number of pack years and histological disease severity
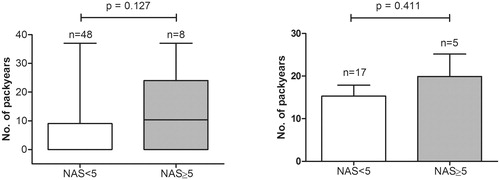
Overall, patients with NAS <5 compared to patients with NAS ≥5 had a median number of pack years of, respectively, 0 (IQR 0–9) versus 10.3 (IQR 0–24) (p = .127) (). There were no differences in clinical features, including age, sex, BMI, the presence of DM and alcohol use between both groups (data not shown). Also after exclusion of never smokers, no statistical difference could be observed in number of pack years (NAS <5; 15.3 ± 10.6 vs. 19.9 ± 11.7 in NAS ≥5; p = .411) (). There was no correlation between number of pack years and NAS in ever smokers (Pearson’s R = 0.072, p = .752).
Figure 2. Number of pack years in patients with a NAS lower than 5 compared to patients with a NAS of 5 or higher. Panel A shows the difference in number of pack years (represented with boxplots with median, IQR and min–max value) for the whole cohort. Panel B shows the difference in number of pack years (represented as mean + standard error of the mean) for ever smokers (active and past smokers).
Discussion
Smoking and fatty liver disease place a heavy burden on health care systems worldwide. This study addresses the possible synergistic effect of smoking on histological outcomes of nonalcoholic fatty liver disease.
We demonstrate in this study that NAFLD patients with advanced fibrosis smoked more pack years than patients with no or early fibrosis. These results corroborate the association between smoking and severity of NAFLD fibrosis as previously shown [Citation7]. In a smaller study, no association between smoking and severity of fibrosis in NAFLD patients was observed, which may be the result of patient numbers, comorbidity and selection [Citation8].
The underlying pathomechanisms of our observed association cannot be clarified with our cross-sectional cohort study design. We can only speculate through which mechanisms smoking aggravates fibrosis in NAFLD, based on previous experimental research.
First, the presence of inflammation as a risk factor for progression to fibrosis in NAFLD has been widely shown [Citation10]. However, we did not find an association between smoking and NASH or severity of inflammation, ballooning and steatosis. An increasing number of pack years was not correlated with a higher NAFLD activity score (NAS). The NAS did not differ between never and ever smokers. Similar results were seen in other studies [Citation7,Citation8].
The heterogeneity of NAFLD could be a possible reason for this, with many environmental and genetic factors influencing disease course [Citation11]. These factors could have been present in our study cohort and are impossible to correct for. Furthermore, our cohort contained only a small percentage (14%) of patients with active steatohepatitis and could therefore be underpowered to show a possible association between smoking and histological disease severity.
Second, also a direct effect on fibrogenesis from smoking could be postulated as an underlying mechanism. NAFLD is a heterogeneous disease and probably progression is caused by a multi-hit model [Citation12]. Smoking probably can interfere in several pro-inflammatory and pro-fibrotic pathways in experimental NAFLD. Chronic hypoxia, as a consequence of smoking, not only led to more hepatic inflammation but also an increase of pro-fibrotic cytokines [Citation13,Citation14]. Other pathways involved in fibrogenesis, such as more profound lipid accumulation, oxidative stress and insulin resistance, have been shown [Citation15]. Stimulation of the nicotinic acetylcholine receptors in hepatic stellate cells by nicotine in vitro suggests a direct effect of smoking on fibrogenesis [Citation16].
In addition, in other chronic liver diseases such as hepatitis C and primary biliary cholangitis, associations between smoking and fibrosis have been described [Citation17–22].
It is furthermore suggested that smoking increases progression of fibrosis in other organ systems, including the pancreas, kidneys, lungs and heart [Citation23–26].
In our study, patients with advanced fibrosis were older than those with early fibrosis but the number of pack years was not correlated with the age of patients. Therefore, age seems to play a minor role in the association between smoking and fibrosis severity. Nonetheless, it endorses the advice to cease smoking for all patients, irrespective of their biological age.
The retrospective set-up of a small cohort is a major limitation. With extensive chart review and direct questioning of patients on smoking history, we aimed to characterize the cohort as good as possible. However, smoking data were not validated by direct measurement of serum nicotine concentrations, so the possibility of bias by patients is present.
In conclusion, in this study, the number of pack years is associated with severity of liver fibrosis but not with histological disease severity in patients with NAFLD. The observed data may serve as pilot for larger cohorts. These findings still emphasize the recommendation to cease smoking for all patients with NAFLD.
Disclosure of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. 2015;62:S47–S64.
- Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321.
- Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology. 2012;142:711–725e6.
- Machado MV, Diehl AM. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology. 2016;150:1769–1777.
- Tilg H, Moschen AR, Roden M. NAFLD and diabetes mellitus. Nat Rev Gastroenterol Hepatol. 2017;14:32--42.
- Azzalini L, Ferrer E, Ramalho LN, et al. Cigarette smoking exacerbates nonalcoholic fatty liver disease in obese rats. Hepatology. 2010;51:1567–1576.
- Zein CO, Unalp A, Colvin R, et al. Smoking and severity of hepatic fibrosis in nonalcoholic fatty liver disease. J Hepatol. 2011;54:753–759.
- Yilmaz Y, Yonal O, Kurt R, et al. Cigarette smoking is not associated with specific histological features or severity of nonalcoholic fatty liver disease. Hepatology. 2010;52:391.
- Hjelkrem M, Stauch C, Shaw J, et al. Validation of the non-alcoholic fatty liver disease activity score. Aliment Pharmacol Ther. 2011;34:214–218.
- Wree A, Broderick L, Canbay A, et al. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat Rev Gastroenterol Hepatol. 2013;10:627–636.
- Satapathy SK, Sanyal AJ. Epidemiology and natural history of nonalcoholic fatty liver disease. Semin Liver Dis. 2015;35:221–235.
- Day CP, James OF. Steatohepatitis: a tale of two “hits”?. Gastroenterology. 1998;114:842–845.
- Savransky V, Bevans S, Nanayakkara A, et al. Chronic intermittent hypoxia causes hepatitis in a mouse model of diet-induced fatty liver. Am J Physiol Gastrointest Liver Physiol. 2007;293:G871–G877.
- Moon JO, Welch TP, Gonzalez FJ, et al. Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G582–G592.
- Bailey SM, Mantena SK, Millender-Swain T, et al. Ethanol and tobacco smoke increase hepatic steatosis and hypoxia in the hypercholesterolemic apoE(-/-) mouse: implications for a “multihit” hypothesis of fatty liver disease. Free Radic Biol Med. 2009;46:928–938.
- Soeda J, Morgan M, McKee C, et al. Nicotine induces fibrogenic changes in human liver via nicotinic acetylcholine receptors expressed on hepatic stellate cells. Biochem Biophys Res Commun. 2012;417:17–22.
- Zein CO, Beatty K, Post AB, et al. Smoking and increased severity of hepatic fibrosis in primary biliary cirrhosis: a cross validated retrospective assessment. Hepatology. 2006;44:1564–1571.
- Corpechot C, Gaouar F, Chrétien Y, et al. Smoking as an independent risk factor of liver fibrosis in primary biliary cirrhosis. J Hepatol. 2012;56:218–224.
- Pessione F, Ramond MJ, Njapoum C, et al. Cigarette smoking and hepatic lesions in patients with chronic hepatitis C. Hepatology. 2001;34:121–125.
- Tsochatzis E, Papatheodoridis GV, Manolakopoulos S, et al. Smoking is associated with steatosis and severe fibrosis in chronic hepatitis C but not B. Scand J Gastroenterol. 2009;44:752–759.
- Altamirano J, Bataller R. Cigarette smoking and chronic liver diseases. Gut. 2010;59:1159–1162.
- Liu B, Balkwill A, Roddam A, et al. Separate and joint effects of alcohol and smoking on the risks of cirrhosis and gallbladder disease in middle-aged women. Am J Epidemiol. 2009;169:153–160.
- van Geenen EJM, Smits MM, Schreuder TCMA, et al. Smoking is related to pancreatic fibrosis in humans. Am J Gastroenterol. 2011;106:1161–1166.
- Sekhon HS, Proskocil BJ, Clark JA, et al. Prenatal nicotine exposure increases connective tissue expression in foetal monkey pulmonary vessels. Eur Respir J. 2004;23:906–915.
- Goette A, Lendeckel U, Kuchenbecker A, et al. Cigarette smoking induces atrial fibrosis in humans via nicotine. Heart. 2007;93:1056–1063.
- Zhang G, Kernan KA, Thomas A, et al. A novel signaling pathway: fibroblast nicotinic receptor alpha1 binds urokinase and promotes renal fibrosis. J Biol Chem. 2009;284:29050–29064.