
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Background
Familial Pancreatic Cancer (FPC) is responsible for up to 10% of all cases of pancreatic ductal adenocarcinoma (PDAC). Individuals predisposed for FPC have an estimated lifetime risk of 16–39% of developing PDAC. While heritability of PDAC has been estimated to be 36% in a Nordic twin study, no heritability estimate specific on FPC has been reported.
Methods
A national cohort of Danish families with predisposition for FPC is currently included in a screening program for PDAC at Odense University Hospital. Family members included in the screening program were interviewed for pedigree data including: cases of PDAC among first-degree relatives (FDRs) and number of affected/unaffected siblings. Heritability for FPC in the predisposed families was assessed by doubling the estimated intra-class correlation coefficient (ICC) from a random intercept logistic model fitted to data on FDRs.
Results
Among families with predisposition for FPC, 83 cases of PDAC were identified. The median age at diagnosis of PDAC was 66 years, and median time from diagnosis to death was 7.6 months. A total of 359 individuals were found as unaffected FDRs of the 83 PDAC cases. The retrieved FDRs included a total of 247 individuals in sibship and 317 individuals in parent-offspring relatedness. We estimated an ICC of 0.25, corresponding to a narrow sense additive heritability estimate of 0.51 in the FPC family cohort.
Conclusion
We have established a nation-wide cohort of FPC families to facilitate clinical and genetic studies on FPC. The estimated heritability of 51% prominently underlines a strong genetic background of FPC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is currently the seventh most fatal cancer type worldwide and has a 5-year survival of around 5% across both high- and low-income countries [Citation1,Citation2]. The incidence of PDAC is increasing globally along with mortality rates – especially prominent in high-income countries, thereby posing a substantial burden of life lost to disease in western populations [Citation3]. While the increase in the incidence of sporadic PDAC may be attributed to a rising prevalence of obesity, diabetes and life-style related comorbidities, it is estimated that up to 10% of all PDAC cases are caused by hereditary factors, including hereditary cancer syndromes and familial pancreatic cancer (FPC) [Citation4].
The heritability of PDAC has been a long-discussed topic with an array of epidemiological studies showing family history to be an important risk factor for developing the disease [Citation5]. The definition of FPC was firstly introduced in 1998 as being presence of PDAC among at least 2 first degree relatives (FDR) in the same family [Citation6]. Predisposition for FPC contributes a largely increased risk of developing PDAC among all FDRs in the affected family [Citation4,Citation7]. The life-time risk of FPC predisposed individuals ranges from 16% to 39% depending on the number of FDRs with PDAC. In Denmark, the incidence rate of PDAC is around 900 cases per year, with 70–90 of the yearly cases estimated to be attributed to hereditary factors [Citation8].
Because of the low incidence of FPC and resulted difficulty in sample collection, familial aggregation is vaguely assessed. Currently, the familial correlation of PDAC has been investigated by a consortium study including Nordic twins from Sweden, Denmark and Finland reporting a low heritability estimate of 0.36 [Citation9]. Even with pooled samples (involving only 4 concordant twin pairs), the estimate is borderline significant with a 95% confidence interval of 0–0.53. A more recent study on Nordic twins estimated the risk of PDAC for co-twins as 4.3% (1.5–11.6%) for MZ and 3.7% (1.5–8.6%) for DZ pairs, furtherly suggesting a genetic component in pancreatic cancer [Citation10]. Based on single-nucleotide polymorphism (SNP) genotype data in a genome-wide association study (GWAS), a downward heritability estimate of 0.21 was reported due to limited genetic contribution covered by the available SNPs [Citation11]. The reported heritability estimates using twins or SNPs data were for pancreatic cancer in general – and not for FPC in particular. This means that these estimates do not represent the genetic contribution to FPC, which accounts for only a small proportion of all forms of pancreatic cancer. As an alternative, the family design can be used to assess the heritability of diseases or health-related phenotypes. In family studies, it is often easier to recruit participants because of increased number of samples meeting eligibility criteria. As full siblings and parent-offspring units (first-degree relatives, FDRs) are expected to share 50% of their genetic material, additive heritability can be estimated by doubling the correlation on disease status (concordance and discordance rate), calculated using pedigree data from FPC families [Citation12].
This paper presents a nationwide cohort of FPC families collected in a long-term screening program and uses the family design to assess the genetic contribution to the disease by estimating correlation on FPC in FDRs. Using a unique cohort from the Danish population, this study aims at providing a novel assessment of the heritability estimate specific for FPC.
Materials and methods
Definition of FPC and inclusion in screening program
A national cohort of 27 large Danish families with disposition for FPC is currently included in a screening program for PDAC at the Department of Medical Gastroenterology, Odense University Hospital, Denmark.
Prior to inclusion in the screening program, each family was diagnosed for a predisposition to FPC at the department of clinical genetics in a regional hospital [Citation13]. Based on previous definitions at our institution and on international consensus criteria [Citation14], familial predisposition for FPC was defined as the presence of either: (1) Two FDRs with PDAC, with at least one of the cases debuting <50 years; or (2) at least three FDRs with PDAC.
The current screening program was initiated in 2007 in a collaborative effort by the five departments of clinical genetics in Denmark (situated at: Odense University Hospital, Aarhus University Hospital, Aalborg University Hospital, Vejle Hospital – Sygehus Lillebaelt and Rigshospitalet) and Odense Pancreas Center (OPAC), Odense University Hospital. Individuals at risk (i.e. FDRs to PDAC cases in the FPC families) are offered inclusion in the screening program after reaching an age corresponding to 5 years before the age of the patient with the earliest diagnosis of PDAC in the family; but no later than at 50 years of age. Screening for PDAC of individuals at risk includes yearly imaging (endoscopic ultrasonography with fine needle biopsy, when relevant) of the pancreas along with PDAC blood markers (e.g. Cancer antigen 19-9, CA19-9) – with the possibility to individualize the time interval between screening sessions if needed, in concordance to the ‘International Cancer of the Pancreas Screening (CAPS) Consortium’ Guidelines [Citation15].
Family members included in the screening program were interviewed and the following data were obtained: cases of PDAC among FDRs (e.g. familial relation, age at diagnosis of PDAC), number of affected/unaffected siblings. Data were validated and supplemented with pedigrees for each FPC family obtained from the relevant departments of clinical genetics in Denmark.
Currently, genetic testing has only been performed on a very limited amount of PDAC cases and their FDRs. The indication for genetic testing for BRCA-1/2 mutations was based on assessment by the local department of clinical genetics that performed the initial genetic counselling of the respective families – prior to referral of each family to our institution for inclusion in the screening program. The criteria for genetic testing were based on local guidelines at each institution, and testing for BRCA-1/2 was mainly performed in families in suspicion of having aggregation of BRCA related cancers (e.g. breast and ovarian cancer). As the gene tests were not done specifically for PDAC, limited information on BRCA-1/2 mutations is available for the FPC cohort. Nonetheless, the pedigree structure of the FPC family cohort allows segregation analysis of the available mutation data to assess their pathogenicity in FPC. For each mutation, a Cosegregation Likelihood Ratio (CSLR) as proposed by Mohammadi and colleagues [Citation16] was estimated, with CSLR > 1 indicating increased likelihood that the mutation is pathogenic and CSLR < 1 indicating likelihood that a variant is benign.
Statistical analysis
In order to estimate the heritability of FPC in our samples, we first estimated the intra-class correlation coefficient (ICC) derived from the random intercept logistic model [Citation17,Citation18]. The random intercept logistic model is given by:
Here bi is the random effect of FDR cluster i. It is assumed that bi follows a normal distribution with mean 0 and variance σ2 (between cluster variance). This model can be viewed as a latent-response model where Yij=1 if
and 0 otherwise.
follows a logistic distribution with mean 0 and variance π2/3.
ICC is defined as the ratio of between-cluster variance to the total variance and calculated as where v2 is a sample estimate of between-cluster variance σ2, and the denominator is the total variance, which is the sum of between-cluster variance (v2) and within-cluster variance (π2/3) [Citation18]. For the fixed total variance observed, the higher the proportion of between-cluster variance, the lower the proportion of within-cluster variance which means higher correlation of samples within each cluster (family). The narrow sense additive heritability is then estimated as h2 = 2*ICC. The 95% confidence intervals for the estimated ICC and h2 can be obtained by bootstrapping resampling. The random intercept logistic model was fitted using the glmer() function of the lme4 package in R (https://www.r-project.org/).
The Cox proportional hazard model was fitted to estimate the effects of selected variables on survival of PDAC patients after diagnosis. The fitting of Cox model took family relatedness into account by setting families as clusters. The functions coxph() and Surv() from the R package survival were used to perform survival analysis.
Ethics
Data collection from patient interviews and pedigree charts was conducted with the approvals from the Danish National Committee on Health Research Ethics (NVK) (project number: 1604008) and the Danish Data Protection Agency (project number: 18/54160).
Results
Basic description of samples
A total of 63 individuals from 27 families with predisposition for FPC currently included in the screening program were interviewed for pedigree data on cases of FPC among FDRs and number of affected/unaffected siblings. Data from the patient interviews were validated with pedigree charts for the 27 families obtained from the relevant departments of clinical genetics.
In total, 1567 samples – consisting of 83 PDAC cases and 1484 unaffected individuals from 27 families with predisposition for FPC were included. For all families, demographical data of all known individuals with FPC were registered along with data on their FDRs. Detailed information about pedigree size and number of affected siblings and parents by each family can be found in Supplementary Table S1. Pedigree sizes varied from 9 to 148 individuals with a median size of 50 (). Among the 1567 samples, 359 individuals (195 males, 164 females) were found as unaffected FDRs of the 83 PDAC cases (38 males, 45 females). Proportion of the PDAC cases among FDRs was 16% (n = 38/233) in males, 22% (45/209) in females, and 19% (83/442) in all samples, showing no sex difference (p = .14). Comparison of FDRs between PDAC and unaffected groups are shown in . Seventy-two out of the 83 PDAC cases and 175 out of the 359 unaffected FDRs were in sibship, with sibship size ranging from 2 to 17 (median: 5) individuals (). Eighty-two out of the 83 PDAC cases and 235 out of the 359 unaffected FDRs were in parents-offspring relatedness (). Median age at diagnosis of PDAC was 66 years (range: 34–96 years) with no sex difference (t = 1.77, p = .08) ( and ). The median age at death for all PDAC patients was 67 years (range: 35–96 years), 62 years (range: 35–96) for males and 69 years (range: 40–87) for females ( and ).
Figure 1. Histogram for the distribution of pedigree size (a), sibship size (b) and parent-offspring relatedness (c,d).
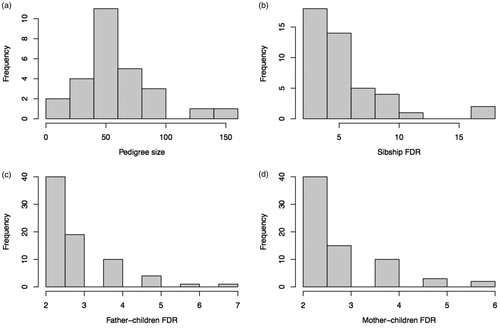
Figure 2. Histogram for median age at PDAC diagnosis (a) and median age at death (b). Kaplan–Meier plot for age at death (c) and survival time from diagnosis to death (d), for male (blue) and female (red) PDAC patients.
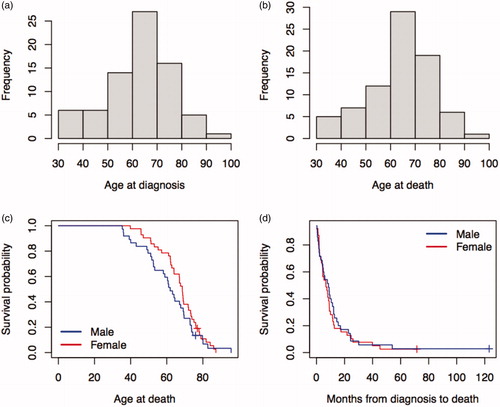
Table 1. Comparison of FDRs between PDAC and unaffected groups.
Table 2. Age at diagnosis and age at death of PDAC patients.
Multiple carriers of family-specific mutations have been found in four families (family numbers 6, 8, 13 and 26) in BRCA-2 and none in BRCA-1. For segregation analysis, we trimmed the large pedigrees (in families 6 and 13) to eliminate uninformative branches. Segregation analysis on the four families (Supplementary Figure S1) estimated CSLRs of 2.48 for family 6 and 1.32 for family 8 suggesting that the corresponding BRCA-2 mutations are pathogenic for PDAC. For families 13 and 26, we obtained CSLRs < 1 (0.17 for family 13 and 0.54 for family 26) indicating the corresponding mutations are benign for PDAC.
Survival of PDAC patients
The Kaplan-Meier plot showed no sex difference in age at death for PDAC patients (Cox model coef. 0.25, p = .27) (). In , the sex-stratified Kaplan-Meier survival curves indicated no difference in survival from diagnosis to death between male and female PDAC cases (Cox model coef. −0.1, p = .66). The mean survival time was 8.5 months for males and 6.7 months for females with an overall mean of 7.6 months (). Two cases survived over 60 months () resulting in a low 5-year survival rate of 2.5%.
Heritability estimate
A random intercept logistic model was fitted with PDAC as outcome and FDR as a random effect variable or cluster. We estimated a random effect variance of 1.12 and an ICC of 0.25. The 95% confidence interval (CI) for the ICC estimate was obtained using bootstrap re-sampling (5000 replicates). In Supplementary Figure S2, we show the distribution of bootstrap ICCs with a median value of 0.25 and a 95% CI: 0.17–0.33. The histogram displays a typical symmetric normal distribution for the bootstrapping ICCs with a median exactly equal to the estimated ICC (0.25) – indicating stably high performance of the random intercept logistic model in fitting the data. The narrow sense heritability, 2*ICC, was calculated to 51% with corresponding 95% CI: 0.35–0.66.
Discussion
We have established a nation-wide cohort of FPC families in a Scandinavian population as a useful resource for clinical and basic research of FPC, especially valuable for exploring the genetic architecture of the disease. Our analysis of clinical features shows that FPC is characterized by high lethality, with a low 5-year survival rate of 2.5% from the cohort and high familial clustering of around 19% of occurrence among FDRs in the respective families, with the latter implying a strong genetic component in FPC. In a US study, 270 pancreatic cancer cases were observed in 2238 FDRs (247 probands and 23 FDR cases) accounting for 12% [Citation19]. The proportion is lower than our estimate because the families were not ascertained using the FPC criteria as in our study. In a recent German study, 374 FPC cases were observed in 1686 family members with an estimated FPC rate of 22% [Citation20]. Statistical testing showed that their estimate is not significantly different from ours (chi-squared = 2.21, p = .14).
To our knowledge, this study provides the first heritability estimate on FPC. Because of the rarity of FPC, estimation of the genetic contribution to the disease using genetically related individuals has been difficult. This study applies a mixed effect modeling to handle relatedness – enabling the estimation of ICC as a traditional correlation in relative pairs (e.g. paired individuals in sibship or in parent-offspring relatedness). For example, the mixed modeling allows the inclusion of any sibship size, unlimited to sibling pairs, which is a different concept to the traditional correlation. By specifying relatedness in a family, parents and children relationship can also be included in the mixed model. The flexibility of the mixed model enables the use of all FDRs in a family for estimating ICC. Our analysis obtained a heritability estimate of 51% – which is much higher than the previous estimates of 21% in a GWAS study [Citation11] and 36% in a Nordic twin study [Citation9] respectively. It needs to be mentioned that the reported estimates using GWAS and twins were based on all types of pancreatic cancer, although PDAC cases among concordant twin pairs may fulfill the FPC criteria [Citation21].
Our heritability estimate was calculated using a cohort of FPC cases and FDRs in Danish families with predisposition for FPC. It is estimated that people with 2 FDRs with FPC have a 6.4-fold higher risk for developing the disease, while those with at least 3 FDRs with FPC have a 32-fold risk [Citation22]. High risk of family history for pancreatic cancer was also reported by a meta-analysis [Citation5]. These data support our heritability estimate, indicating a high genetic contribution to the development of FPC.
The calculated heritability using ICC represents a narrow sense additive genetic model. The estimated heritability is a narrow sense because it captures only the proportion of genetic variations that are due to additive genetic effects (i.e. dominant or recessive inheritance was not taken into account). Multiple environmental exposures within families such as heavy smoking, alcohol, obesity, etc. have been reported as risk factors for early onset of precursor lesions of pancreatic cancer [Citation23,Citation24]. Environmental factors may be especially relevant to FPC as these patients develop more precursor lesions with differential gene expression patterns compared to pancreatic cancer patients without familial predisposition [Citation25–27]. Accumulative environmental exposures such as drinking and smoking confer a strong risk of pancreatic inflammation (i.e. pancreatitis) [Citation28], while inflammatory signals have been shown to implicate both development of precursor lesions and progression to PDAC [Citation29]. This means that sharing of the risky environmental factors within families may also contribute to family aggregation of FPC. Hence, the ICC-based approach provides an upper bound estimate of heritability. Findings of the present study emphasize the genetic component in pancreatic cancer (PC) as reflected by reports of previous population-based studies – indicating an increased risk of PDAC among FDRs to patients with PC in general [Citation5,Citation30,Citation31].
This study estimated the heritability of FPC based on PDAC phenotype correlation among FDRs to assess the overall genetic contribution to FPC without using genomic information. This is similar to the traditional twin studies that estimate heritability based on phenotype correlation in twin pairs with no molecular data needed. In genetic epidemiology, a moderate to high heritability estimate calls for the practice to explore the genetic architecture of the disease using genetic association studies. The estimated heritability of 51% for FPC highlights the importance of investigating genetic variations underlying the etiology of the disease. While the phenotype-based heritability estimate provides an overall assessment of genetic contribution to FPC, genetic association studies including GWAS based on genomic information can help to identify the DNA sequence variations associated with FPC. A GWAS conducted on patients with family history of pancreatic cancer or diagnosed under age 50 reported multiple SNPs influencing risk of PDAC and other cancers [Citation32]. However, it is now widely known that the common genetic variants detected by GWAS only explain a limited proportion of the estimated overall genetic contribution (heritability) to a specific complex disorder – a phenomenon referred to as the ‘missing heritability’ problem, and thus rare variants association analysis has been proposed as an alternative to GWAS to dissolve the issue [Citation33].
Presently, an increasing amount of next generation sequencing (NGS) studies on FPC are being performed – with previous studies identifying 11 PDAC susceptibility genes, including prominent gene mutations in BRCA-1/2, PALB2, CDKNA2 and ATM being associated with FPC [Citation34,Citation35]. Nonetheless, only about 12% of all FPC cases carry any of these mutations – meaning that the germline-component of >80% of all FPC cases still remains unknown [Citation36]. Given our heritability estimate, more genome-scale analyses are called for in order to uncover the underlying genetic architecture of FPC.
One important application of the family cohort is for assessing the effects of detected mutations on FPC development. Although with limited data on genetic testing, our application of segregation analysis on BRCA-2 mutations exemplifies the utility of the cohort for future genetic studies. With more gene testing and whole-genome sequencing to be performed in the cohort, segregation analysis will help with evaluating the pathogenicity of detected unclassified variants. Such information will be useful for FPC-related basic research and clinical practice.
A limitation of this study is the age structure of the samples – as the age-at-onset of FPC cases may affect the estimate. The FDRs in our analysis included older siblings of each parent, as well as siblings of the offspring generation (i.e. the youngest generation of a pedigree) – meaning that FDRs in the youngest generation of each pedigree may have not expressed the disease yet. Although the year of birth was adjusted for in our estimation of ICC, the heritability could still have been underestimated in our study. Nevertheless, the observed age-at-onset of FPC patients in the present study (mean age: 63 years; median: 66 years) is very similar to reports from previous studies (mean age: 64–65 years) [Citation37] – being significantly younger than the age-at-onset of sporadic pancreatic cancer (median age: 71 years) [Citation38]. Another limitation is the relatively small sample size available to us in this study, preventing us from stratifying heritability estimates by sex. Future large-scale studies on FPC family cohorts could help verifying our estimates and examine potential differences between genders in FPC heritability.
Heritability studies are an important part of traditional genetic epidemiology for assessing the genetic basis in disease development. Our application of the mixed logistic regression model for estimation of ICC and heritability serves as a novel approach that promotes mixed modelling in assessing and measuring the genetic contribution to familial cancers.
Conclusion
The present study is the first to assess a heritability estimate for FPC – using a nation-wide cohort of FPC families in a Scandinavian population. The estimated additive heritability of 51% prominently underlines a strong genetic contribution to the development of FPC, which encourages mapping of the underlying genetic variants using high-throughput next generation sequencing (NGS) analysis. The established family cohort presented in this study is a unique resource for clinical and NGS studies of FPC currently underway.
Author contributions
MT, OBSM and MTJ formulated and designed the study. MT collected the data and conducted statistical analysis and interpretation. MT drafted the paper. OBSM, MTJ, KB, AMG, MBM and SD revised the paper. All authors approved the final version of the manuscript to be published.
| Abbreviations | ||
| CI | = | confidence interval |
| FDR | = | first degree relative |
| FPC | = | familial pancreatic cancer |
| ICC | = | intra class correlation coefficient |
| NGS | = | next generation sequencing |
| NVK | = | Danish National Committee on Health Research Ethics |
| OPAC | = | Odense Pancreas Center |
| PC | = | pancreatic cancer |
| PDAC | = | pancreatic ductal adenocarcinoma |
Supplemental Material
Download PDF (4.6 KB)Supplemental Material
Download PDF (111.3 KB)Supplemental Material
Download MS Word (15.2 KB)Acknowledgements
The authors want to issue a special thanks to the following hospital departments for their contribution with relevant pedigrees of the included FPC families in this study: the Department of Clinical Genetics, Aalborg University Hospital, Aalborg, Denmark; the Department of Clinical Genetics, Aarhus University Hospital, Aarhus, Denmark; the Department of Clinical Genetics, Odense University Hospital, Odense, Denmark; the Department of Clinical Genetics, Rigshospitalet, Copenhagen, Denmark; the Department of Clinical Genetics, Vejle Hospital – Sygehus Lillebaelt, Vejle, Denmark.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- The global, regional, and national burden of pancreatic cancer and its attributable risk factors in 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2019;4(12):934–947.
- McGuigan A, Kelly P, Turkington RC, et al. Pancreatic cancer: a review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol. 2018;24(43):4846–4861.
- Carrato A, Falcone A, Ducreux M, et al. A systematic review of the burden of pancreatic cancer in europe: real-world impact on survival, quality of life and costs. J Gastrointest Cancer. 2015;46(3):201–211.
- Petersen GM. Familial pancreatic adenocarcinoma. Hematol Oncol Clin North Am. 2015;29(4):641–653.
- Permuth-Wey J, Egan KM. Family history is a significant risk factor for pancreatic cancer: results from a systematic review and meta-analysis. Fam Cancer. 2009;8(2):109–117.
- Hruban RH, Petersen GM, Ha PK, et al. Genetics of pancreatic cancer. From genes to families. Surg Oncol Clin N Am. 1998;7(1):1–23.
- Brune KA, Lau B, Palmisano E, et al. Importance of age of onset in pancreatic cancer kindreds. J Natl Cancer Inst. 2010;102(2):119–126.
- Weble TC, Bjerregaard JK, Kissmeyer P, et al. Incidence of pancreatic cancer in Denmark: 70 years of registration, 1943-2012. Acta Oncol. 2017;56(12):1763–1768.
- Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78–85.
- Mucci LA, Hjelmborg JB, Harris JR, et al. Familial risk and heritability of cancer among twins in Nordic countries. JAMA. 2016;315(1):68–76.
- Chen F, Childs EJ, Mocci E, et al. Analysis of heritability and genetic architecture of pancreatic cancer: a PanC4 study. Cancer Epidemiol Biomarkers Prev. 2019;28(7):1238–1245.
- Tenesa A, Haley CS. The heritability of human disease: estimation, uses and abuses. Nat Rev Genet. 2013;14(2):139–149.
- Jorgensen MT, Mortensen MB, Gerdes AM, et al. Familial pancreatic cancer. Scand J Gastroenterol. 2008;43(4):387–397.
- Joergensen MT, Gerdes AM, Sorensen J, et al. Is screening for pancreatic cancer in high-risk groups cost-effective? – experience from a Danish national screening program. Pancreatology. 2016;16(4):584–592.
- Goggins M, Overbeek KA, Brand R, et al. Management of patients with increased risk for familial pancreatic cancer: updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut. 2020;69(1):7–17.
- Mohammadi L, Vreeswijk MP, Oldenburg R, et al. A simple method for co-segregation analysis to evaluate the pathogenicity of unclassified variants; BRCA1 and BRCA2 as an example. BMC Cancer. 2009;9:211.
- Moineddin R, Matheson FI, Glazier RH. A simulation study of sample size for multilevel logistic regression models. BMC Med Res Methodol. 2007;7:34.
- Wu S, Crespi CM, Wong WK. Comparison of methods for estimating the intraclass correlation coefficient for binary responses in cancer prevention cluster randomized trials. Contemp Clin Trials. 2012;33(5):869–880.
- Schenk M, Schwartz AG, O'Neal E, et al. Familial risk of pancreatic cancer. J Natl Cancer Inst. 2001;93(8):640–644.
- Bartsch DK, Matthäi E, Mintziras I, et al. Characteristics of pure familial pancreatic cancer families and those with additional breast cancer. OAJOM. 2020;4(1):339–346.
- Benzel J, Fendrich V. Familial pancreatic cancer. Oncol Res Treat. 2018;41(10):611–618.
- Brand RE, Lerch MM, Rubinstein WS, et al. Advances in counselling and surveillance of patients at risk for pancreatic cancer. Gut. 2007;56(10):1460–1469.
- Benzel J, Fendrich V. Chemoprevention and treatment of pancreatic cancer: update and review of the literature. Digestion. 2018;97(4):275–287.
- Olson SH, Kurtz RC. Epidemiology of pancreatic cancer and the role of family history. J Surg Oncol. 2013;107(1):1–7.
- Rustgi AK. Familial pancreatic cancer: genetic advances. Genes Dev. 2014;28(1):1–7.
- Humphris JL, Johns AL, Simpson SH, et al. Clinical and pathologic features of familial pancreatic cancer. Cancer. 2014;120(23):3669–3675.
- Tan M, Schaffalitzky de Muckadell OB, Joergensen MT. Gene expression network analysis of precursor lesions in familial pancreatic cancer. J Pancreat Cancer. 2020;6(1):73–84.
- Yadav D, Whitcomb DC. The role of alcohol and smoking in pancreatitis. Nat Rev Gastroenterol Hepatol. 2010;7(3):131–145.
- Steele CW, Jamieson NB, Evans TRJ, et al. Exploiting inflammation for therapeutic gain in pancreatic cancer. Br J Cancer. 2013;108(5):997–1003.
- Shirts BH, Burt RW, Mulvihill SJ, et al. A population-based description of familial clustering of pancreatic cancer. Clinical gastroenterology and hepatology: the official clinical practice journal of the. Am Gastroenterol Assoc. 2010;8(9):812–816.
- Kohli DR, Smith KR, Wong J, et al. Familial pancreatic cancer risk: a population-based study in Utah. J Gastroenterol. 2019;54(12):1106–1112.
- Childs EJ, Chaffee KG, Gallinger S, et al. Association of common susceptibility variants of pancreatic cancer in higher-risk patients: A PACGENE study. Cancer Epidemiol Biomarkers Prev. 2016;25(7):1185–1191.
- Zuk O, Schaffner SF, Samocha K, et al. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci U S A. 2014;111(4):E455–64.
- Roberts NJ, Norris AL, Petersen GM, et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. 2016;6(2):166–175.
- Zhen DB, Rabe KG, Gallinger S, et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med. 2015;17(7):569–577.
- Chaffee KG, Oberg AL, McWilliams RR, et al. Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet Med. 2018;20(1):119–127.
- Petersen GM. Familial pancreatic cancer. Semin Oncol. 2016;43(5):548–553.
- Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144(6):1252–1261.