
Abstract
Introduction
Diffuse peripheral neuropathy is a well-known complication of several conditions, whereas many patients have peripheral neuropathy of unknown etiology and pathophyisology. Increased knowledge of mechanisms may provide insight into enteric neuropathy with gastrointestinal dysmotility. The aim of the present systematic review was to identify mechanisms behind diffuse idiopathic peripheral neuropathies in humans.
Methods
Searches were performed in PubMed, Embase, and Web of Science. Human original and review articles, written in English, describing mechanisms behind diffuse peripheral neuropathy verified by objective examinations were intended to be studied. Articles that described animal models, well-described hereditary diseases, drug-induced neuropathy, pain syndromes, malnutrition, and local neuropathy were excluded.
Results
In total, 4712 articles were identified. After scrutinizing titles and abstracts, 633 remained and were studied in full text. After the removal of articles not fulfilling inclusion or exclusion criteria, 52 were finally included in this review. The most frequently described neuropathy was diabetic neuropathy, with a wide range of mechanisms involving mitochondrial dysfunction such as oxidative stress and inflammation. Microvascular changes in diabetes and vasculitis lead to ischemia and secondary oxidative stress with inflammation. Structural changes in neurons and glial cells are observed, with abnormalities in different neurotrophic factors. Neuropathy induced by autoantibodies or immunological mechanisms is described in infectious and systemic inflammatory diseases. Several ion channels may be involved in painful neuropathy. No study identified why some patients mainly develop large fiber neuropathy and others small fiber neuropathy.
Conclusion
Metabolic and immunological factors and channelopathy may be considered in diffuse idiopathic peripheral neuropathy.
Background
It is well known that several diseases may lead to diffuse peripheral neuropathy in the autonomic nervous system, including the enteric nervous system, and the sensorimotor system [Citation1]. Since the awareness of enteric neuropathy is limited, it may take time before reaching a correct diagnosis as well as proper treatment of the patient. Signs and symptoms of cardiovascular autonomic neuropathy and sensorimotor neuropathy are more often recognized, whereas enteric neuropathy with gastrointestinal (GI) dysmotility may be classified as psychological and functional disorders instead of organic neuropathy [Citation1]. Undiagnosed, enteric neuropathy may lead to malnutrition and, thereby, further neurological deterioration [Citation2].
About two-thirds of patients with diabetes develop any neuropathy and 50% develop enteric neuropathy [Citation3,Citation4]. Diabetes care mainly focuses on glucose homeostasis, blood pressure, and lipid levels. However, GI dysmotility with impaired deliveries of food and drugs may aggravate the dysregulated glucose homeostasis and have great negative impact on the metabolic control and quality of life [Citation1,Citation3]. Autonomic neuropathy is closely associated with GI dysmotility [Citation5], and systemic autoimmune diseases often involve the GI tract [Citation6,Citation7]. Neurological diseases, such as Parkinson’s disease or multiple sclerosis, may start in the GI tract before lesions are observed in the brain [Citation8].
Peripheral neuropathy may depend on axonal or demyelinating neuropathy [Citation3]. Diffuse small nerve fiber neuropathy (SFN) is one of the most common entities and is associated with symptoms from both autonomic and sensory nerves [Citation9]. Approximately 90% of the autonomous nervous system is composed of postganglionic, myelinated Aδ fibers and unmyelinated C fibers, which are also frequently found as sensory nerves in the skin [Citation10]. These small autonomic efferent fibers transmit complex signals from the autonomic ganglia to blood vessels and internal organs to maintain homeostasis through the release of electrical and paracrine chemical signals regulating motility, inflammation, and healing in the tissue and run in parallel with sensory afferent nerves which transmit pain, itch, and temperature from the skin [Citation11].
Many patients have signs and symptoms of peripheral neuropathy without any obvious etiology. In these idiopathic cases, it may be difficult and time-consuming to rule out the causes of neuropathy. Most mechanism studies are performed in animals and need to be confirmed in humans, due to different physiology and anatomical distribution of structures between different species [Citation12,Citation13]. Few reviews about mechanisms behind enteric neuropathy and GI dysmotility or general peripheral neuropathy have been published. The aim of the present systematic review was therefore to identify mechanisms behind diffuse idiopathic peripheral neuropathy involving internal organ systems in humans, and to identify the background to that some patients develop mainly large fiber neuropathy and others mainly SFN.
Material and methods
This review was performed according to the PRISMA (preferred reporting items for systematic reviews and meta-analysis) statement [Citation14], with the help of the PRISMA E & E (explanation and elaboration) document [Citation15]. According to the review protocol, human original and review articles, written in English, which described mechanisms behind diffuse peripheral neuropathy without any known etiology present as large fiber polyneuropathy or SFN were intended to be studied. Articles that only described animal models for the mechanism studies, well-known hereditary diseases, drug-induced neuropathy, malnutrition, pain syndromes, and local damage of peripheral nerves were excluded (). The neuropathy should be verified by objective assessment, e.g., autonomic function tests, assessment of GI motility, skin biopsy, nerve transduction tests, or clinical examination with signs of reduced reflexes and abnormal sensitivity (). Descriptions of neuropathy solely based on subjective symptoms were not accepted. Publications that described correlations of signs and biomarkers or altered concentrations of biomarkers in relation to healthy controls were not included if no causality was described.
Figure 1. Flow chart over the identification and review process.
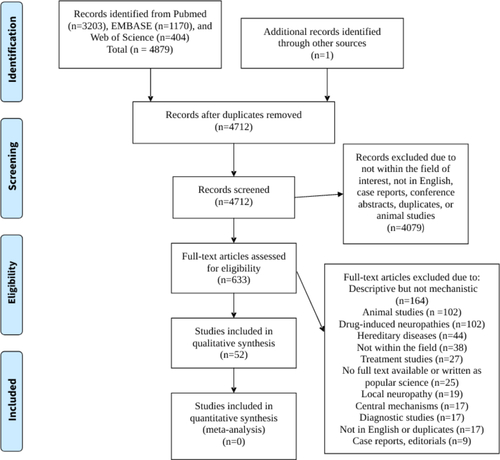
Table 1. Well-known entities of peripheral neuropathy which are not considered in this review.
Information sources
A systematic search strategy was used in three databases: PubMed, Embase (Elsevier), and Web of Science (Clarivate Analytics), on 26 March 2021. The search strategy was designed and executed in collaboration with an information specialist and used keywords and relevant thesaurus terms when applicable. No publication date restrictions were applied, but the language was restricted to English. In Embase, conference abstracts were excluded.
PubMed
The search terms in PubMed were: ((patho*[Title/Abstract] OR patophysio*[Title/Abstract] OR physiopath*[Title/Abstract] OR etiolog*[Title/Abstract]) AND ((‘hypersensitivity’[MeSH Terms]) OR (hyposensitivity[Title/Abstract] OR hypersensitivity[Title/Abstract] OR sensory[Title/Abstract] OR intolerance[Title/Abstract]))) AND ((Peripheral Nervous System Diseases[MeSH Terms]) OR (peripheral neuropathy[Title/Abstract])). The search identified 3203 records.
Embase
The search terms in Embase were: 'peripheral neuropathy’/exp OR 'peripheral neuropathy’:ab,ti AND 'hypersensitivity’/exp OR 'hyposensitivity’/exp OR hypersensitivity:ab,ti OR hyposensitivity:ab,ti OR sensory:ab,ti OR intolerance:ab,ti AND patho*:ab,ti OR patophysio*:ab,ti OR physiopath*:ab,ti OR etiolog*:ab,ti AND NOT 'conference abstract’:it AND [embase]/lim NOT ([embase]/lim AND [medline]/lim). The search strategy identified 404 records.
Web of science
The following topics were used; TOPIC: (peripheral neuropathy) AND TOPIC: ((hypersensitivity OR hyposensitivity)) Refined by: [excluding] DOCUMENT TYPES: (BOOK OR MEETING)
Databases = WOS, BIOSIS, CABI, FSTA, KJD, MEDLINE, RSCI, SCIELO, ZOOREC Timespan = All years, Search language = Auto. These topics rendered 1170 records.
Quality assessment
The screening of all articles was performed by one author (BO). Assessments of full-length articles intended to be included were performed by all authors, independently of each other. In case of discordance, the articles were reread, and after discussion, a consensus was made.
Results
The total search strategy rendered 4879 records, but after deduplication in EndNote, 4711 records in total were identified. One article was found from a reference list. These were screened of titles and abstracts. Animal studies and other studies not intended to be included, articles not written in English, case reports, conference abstracts, and further duplicates were excluded (). After this screening, 633 articles remained which were studied in full length. Further articles were then excluded due to different reasons, most often because mechanisms behind neuropathy were not described (). If an article was not available in full length in any search strategy, the abstract was re-evaluated together and excluded if it was deemed irrelevant for the review or if it was found to be written as popular science. Finally, 52 articles were included in the systematic review ().
Table 2. Studies included in the systematic review.
Hyperglycemia and diabetic neuropathy
Glucose uptake in neurons and Schwann cells is independent on insulin secretion [Citation16]. Hyperglycemia and/or insulin deficiency induces nerve damage in diabetes by several pathways [Citation17], and the damages involve several different tissues and components such as neuronal cells, glial cells, or vascular components [Citation18,Citation19]. Not only manifest diabetes but also the metabolic syndrome, obesity, and pre-diabetes are associated with neuropathy, although the neuropathy is less severe in these entities than in manifest diabetes [Citation20]. Although hyperglycemia has been most often studied, the absolute or relative insulin deficiency per se may contribute to the neuropathy [Citation21].
A cellular pathway of special importance in diabetic neuropathy is the energy-sensing pathway, which consists of the nicotideamine-adenine dinucleotide (NAD+)-dependent Sirtuin 1 (SIRT1)/peroxisome proliferator-activated receptor-γ coactivator α (PGC-1α)/mitochondrial transcription factor A (TFAM or mtTFA) signaling pathway. PGC-1α is involved in the development of diabetes in human subjects and polymorphisms of this gene have been associated with the progression from impaired glucose tolerance to manifest diabetes, and the progression of diabetic neuropathy in animal models [Citation22].
Oxidative stress
The mitochondria are crucial for a long range of biochemical processes such as fatty acid oxidation, nutrient production in the citric acid cycle, oxidative phosphorylation, and ATP production. Hyperglycemia leads to excessive electronic donation to the respiratory chain, in turn resulting in mitochondrial hyperpolarization and increased reactive oxygen species (ROS) production [Citation23]. ROS and reactive nitrogen species (RNS), together classified as RONS, include reactive radical and non-radical derivatives of oxygen and nitrogen from both endogenous and exogenous sources. RONS production induces activities in pathological pathways involved in hyperglycemia-induced oxidative stress: activation of protein kinase C (PKC), increased hexosamine pathway flux, increased production of advanced glycation end products (AGE), and increased polyol pathway flux [Citation21,Citation24]. Further tertiary consequences involve increased levels of mitogen-activated protein (MAP) kinases and decreased levels of neurotrophic support in the form of nerve growth factor (NGF) [Citation16]. Changes in the mitochondria function secondary to oxidative stress and elevated levels of AGE have been linked to neurological diseases and diabetic neuropathy [Citation25]. In one study, increased superoxide anion generation predicted the progression of diabetic neuropathy [Citation26].
Oxidative stress in Schwann cells leads to schwannopathy with direct negative effects on neuronal function in several ways. First, altered function of Schwann cells by hyperglycemia leads to myelin disruption. Second, schwannopathy may lead to reduced axonal propagation by other mechanisms, such as altered ion flux and currents. Third, Schwann cells seem to be involved in peripheral axonal regeneration after traumatic injuries [Citation18].
Impaired mitochondrial buffering of calcium leading to altered Ca2+ currents and increased production of ROS with suboptimal respiratory chain activity in dorsal root ganglion (DRG) neurons have been described in rodents; changes like those in skeletal and cardiac muscle in diabetic human subjects [Citation23,Citation27]. Exercise has been shown to be beneficial in preventing the latter changes in diabetes, and preliminary results show that exercise could reduce pain and increase epidermal fiber density [Citation23].
Atypical sphingolipids, 1-deoxysphingolipids, may represent yet another mechanism and pathway for neurotoxicity since these sphingolipids are elevated in plasma in both type 2 diabetes and hereditary sensory and autonomic neuropathy type 1. Enrichment of a metabolite from these specific sphingolipids in cell experiments results in mitochondrial toxicity [Citation28].
Advanced glycation end products
AGEs are a group of molecules formed when arginine and lysine residues of protein, amino groups on lipids, or guanine nucleic acids become glycated due to sugar exposure. In hyperglycemia, the normal metabolic pathways are overloaded, and excess glucose is shunted into ancillary pathways that become damaging due to increased production of, e.g., AGE. Many steps in these pathways are reversible, but the end products are irreversible and compromise normal neuronal function. Elevated plasma levels of AGE lead to increased accumulation of AGE in different tissues, including different components of peripheral nerves [Citation17,Citation29]. AGE interact with surface receptors, especially the receptor for AGE (RAGE), to induce several intracellular signals and cellular stress [Citation25]. Methylglyoxal, which is a precursor of AGE, has received attention to be a biomarker for predicting the onset and progression of diabetic neuropathy [Citation29].
Polyol pathway and aldose reductase
Hyperglycemia results in increased activity through the polyol pathway, which is one of the most studied pathogenic mechanisms in diabetic neuropathy [Citation21]. High aldose reductase activity (the first enzyme of this pathway) and high polyol pathway activity in experimental animal models result in similar pathological changes in Schwann cells as those observed in humans with diabetic neuropathy [Citation18]. Due to accumulation of the intermediates sorbitol and fructose, increased activity in this pathway leads to further osmotic and oxidative stress and reduced neurotrophic support [Citation21].
Neurotrophic factors
Experimental animal studies have shown that diabetic neuropathy involves specific changes in dorsal root ganglia (DRG) sensory neurons [Citation30]. A greater loss of axonal terminals in the skin of distal parts of the extremities compared with more proximal parts indicates a ‘dying-back’ phenomenon and supports the involvement of DRG also in humans [Citation30]. Both distal symmetric polyneuropathy and SFN are characterized by this dying-back axonopathy, especially in C fibers, and sural nerves from patients with diabetic neuropathy show loss of both myelinated and unmyelinated fibers [Citation31].
Oxidative stress may cause decreased levels of nerve growth factor (NGF) in diabetic skin, which in turn reduces the sensitivity for chemo and heat pain [Citation16,Citation32]. With more advanced diabetic neuropathy, increased levels of messenger RNA for NGF can be detected in keratinocytes, in parallel with upregulated receptors, suggesting compensatory mechanisms. Besides NGF, the expression of messenger RNA for other trophic factors such as brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and glial cell-derived neurotrophic factor (GDNF) have been found to be decreased in the skin of diabetic subjects. It is uncertain whether all changes are primary causal changes or possibly compensatory mechanisms [Citation32]. Finally, also the concentration of a few neuroeffector agents related to NGFs, such as substance p (SP), have been found to be different in diabetes. NGF or the different precursor proteins may regulate the sensitivity and function of small sensory fibers either directly or via these neuroeffector agents [Citation32]. If there is a loss of nerve fibers, each remaining fiber will be hyper-stimulated by an abundance of NGF and other growth factors [Citation33]. However, with more advanced diabetic neuropathy, there is a gradual loss of nerve fibers in parallel with less regenerating fibers [Citation31].
Inflammation
ROS are crucial in several inflammatory processes such as the arachidonic acid cascade and phagocytosis. Both neurons and Schwann cells produce and secrete several cytokines, which are of importance for the degeneration and regeneration of peripheral neurons. Metabolic and microvascular changes affect cytokine production in diabetes, and are involved in the development of peripheral neuropathy [Citation34]. In addition, the higher blood levels of triglycerides, cholesterol, and free fatty acids commonly encountered in diabetic subjects may increase the inflammatory response in Schwann cells. The mechanisms hereby are not completely understood but may involve lipotoxicity of free fatty acids and the uptake of modified LDL via TLRs, in turn increasing the expression of various cytokines and chemokines [Citation18]. Signals from toll-like receptors (TLR) 2 and 4 are dysregulated in human patients with progressive diabetic neuropathy. Further, the TLR-associated genes lipopolysaccharide-binding protein (LBP) and phosphoinositide-3-kinase beta polypeptide (PIK3CB) were dysregulated in prediabetic and diabetic neuropathy across species [Citation35].
Microvascular changes
Microvascular changes are common in diabetes and lead to reduced supply of oxygen and glucose, both by reduced blood flow but also by capillary dysfunction leading to reduced uptake [Citation36].
The microvascular changes may create a hypoxic environment leading to increased inflammation and oxidative stress [Citation18]. On the other hand, the microvascular hypothesis of diabetic neuropathy has been challenged by the fact that sensory neuropathy is the earliest sign of diabetic neuropathy, whereas microvascular changes are likely to involve all neurons. Moreover, diabetic neuropathy can develop prior to microvascular changes [Citation30]. It has thus been hypothesized that disturbances in capillary flow patterns and function, rather than ischemia, is the main responsible mechanism [Citation36]. Thus, microvascular changes may develop in parallel with other causes of diabetic neuropathy [Citation30].
Inflammatory mechanisms in neuropathy
Antibody-mediated neuropathy
A few studies could report mechanisms regarding studies of antibody prevalence in diffuse neuropathy, and those studies described different antibodies in autonomic and sensorimotor neuropathy [Citation37–39]. Contactin-1 (CNTN1) and neurofascin-155 (Nfasc155) are two important cell adhesion molecules involved in the formation of paranodal domains [Citation40]. CNTN1 is also related to contactin-associated protein-1 (Caspr1), which is expressed on the axonal surface and interact with Nfasc155 [Citation41,Citation42]. In a study of patients with chronic inflammatory demyelinating polyradiculopathy (CIDP) of different reasons, 5.5% expressed antibodies against these nodal/paranodal proteins, i.e., Nfasc155, Nfasc140/186, CNTN1, and Caspr1, in comparison to zero in healthy controls and patients with other neuropathological diseases [Citation37]. The presence of antibodies was associated with clinical features and elongated nodes of Ranvier and loss of nodal Nfasc155 and Caspr1 staining in skin biopsies [Citation37]. Sera from sero-positive patients showed reactivity against paranodes and nodes of Ranvier on fibers from murine sciatic nerves [Citation37]. In vitro experiments with human embryonic kidney cells showed that expression of CNTN1/Caspr1 increased the number of cell clusters, whereas the presence of anti-Caspr1 antibodies abolished these clusters [Citation37]. Another CIPD study described macrophage-induced phagocytosis of myelin, with no direct association between autoantibodies and phagocytosis [Citation38]. Specific autoantibodies against the ganglionic nicotinic acetylcholine (ACh) receptor have been identified in panautonomic dysfunction. These autoantibodies caused autonomic failure in animal models and in transfer of maternal antibodies to the child [Citation39].
Gangliosides are membrane-localized sialylated glycosphingolipids present in all cells, but especially abundant in the nervous system, where they occur both in axons and myelin and are highly enriched in the nodes of Ranvier. Glycosphingolipids are involved in the modulation of nociceptive ion channel function in neuropathic pain involving autonomic and sensory Aδ and C fibers. SFN gives rise to pathological pain sensations including intermittent burning pain attacks [Citation43]. Anti-ganglioside antibodies initiate immune attacks against axolemmal gangliosides of myelinated axons causing activation of complements and macrophages [Citation43]. The induced damage disrupt the connection between the axolemma and the myelin loops at the paranodal regions, leading to demyelination and axon degeneration [Citation44].
Immune-mediated neuropathy
The most common HIV-associated peripheral neuropathy is distal sensory polyneuropathy, caused by macrophage-mediated axonal injury due to the release of proinflammatory cytokines and/or lymphocytic infiltration of the perineurium [Citation45]. CIDP is less common, which in the early stages is caused by lymphocyte and macrophage infiltration with complement activation, leading to demyelination. At later stages, remyelination is observed but with the reduction in the density of small myelinated and unmyelinated fibers [Citation45]. The DRG may be involved through several cytokines and other inflammatory mediators such as nitric oxide (NO) and reduced numbers of neurons [Citation45]. It has been indicated that NO-cytotoxicity primarily affects sensory neurons rather than Schwann cells, and that NO exerts direct damage to the axon myelin [Citation46]. A specific micro-RNA, called miR-455-3p, targeting genes involved in encoding factors is of importance for peripheral nerve function, such as NGF. Transfection with this micro-RNA in human neurons leads to reduced beta3-tubulin expression and decreased neural growth [Citation47]. Enteric neuropathy has been described in animal studies but not in human studies [Citation48].
Leprous neuropathy, resulting mainly in sensory loss, is caused by the invasion of M. leprae of Schwann cells. It has been suggested that the nerve damage is mainly due to the immune response, and not the infectious agents per se. Activated CD4+ T-cells that react to presented antigens of Schwann cells cause lysis of the Schwann cells. Moreover, there are interactions of M. leprae with TLR on Schwann cells [Citation49]. Different immunological mechanisms are involved in the nerve damage in different types of leprosy [Citation50]. In leprous skin, reduced levels of NGF may explain the skin hypoalgesia in these patients [Citation32].
The pathological substrate in primary Sjogren’s syndrome (pSS) is ganglionitis mediated by CD8+ T lymphocytes, and the upregulation of proinflammatory cytokines is crucial in the pathophysiology of pSS neuropathy [Citation51]. Peripheral neuropathy occurs in 2%–60% of patients with pSS. The main mechanisms are involvement of vasa nervorum with perivascular inflammatory infiltration, with or without necrosis, leading to ischemia in the tissue and lymphocytic infiltration of DRG [Citation7]. Similar mechanisms leading to ischemia are involved in peripheral neuropathy secondary to vasculitis [Citation6].
Channelopathy in neuropathy
Voltage-gated sodium channels
Voltage-gated sodium channels (VGSCs) are essential in the creation and propagation of the action potential in all excitable cells, including neurons. So far, 9 different variants of alpha-subunits (NaV1.1 – NaV1.9), encoded by SCN1A-SCN5A and SCN8A-SCN11A, have been found in humans with different distribution throughout the nervous system. The variants have different electrophysical properties, and the combination of the different VGSCs in different nerves shape the action potential and the excitability properties of a specific neuronal type [Citation52,Citation53]. Generally, the VGSCs are located in the initial axonal segment, the nodes of Ranvier, and the presynaptic terminals of the peripheral nerves [Citation54]. Gain-of-function of NaV1.7, NaV1.8 and NaV1.9 have been associated with human SFN [Citation55].
NaV1.7, encoded by SCN9A [Citation55], is expressed on various tissues and densely on nociceptors and sympathetic ganglion neurons [Citation56,Citation57]. Gain-of-function variants of this gene are associated with rare painful neuropathic syndromes such as inherited erythromelalgia (IEM) and paroxysmal extreme pain disorder (PEPD), as well as SFN [Citation55]. Almost one-third in a patient cohort with well-defined idiopathic SFN had a genetic gain-of-function with mis-sense variants of the SCN9A, rendering NaV1.7 channel hyperexcitability [Citation57]. The genetic SCN9A variants have been studied in other patients with SFN, and there is a diversity in phenotypic expressions suggesting that the genetic SCN9A variants act as risk factors or amplifiers for SFN. Another potential explanation to the different expressions in genetic variants can be due to other ion channel gene variants [Citation53, Citation55]. Genetic variants of NaV1.7 have also been detected in painful diabetic neuropathy and were associated with channel hyperexcitability [Citation56].
NaV1.8 is expressed in small-diameter primary sensory neurons including DRG, trigeminal ganglia, and nodose ganglia. Gain-of-function mutations of the SCN10A gene that encodes NaV1.8 have been associated with SFN, with and without autonomic dysfunction [Citation9,Citation54,Citation55,Citation58].
NaV1.9 is preferentially expressed in small-diameter DRG neurons and trigeminal neurons, but also in intrinsic myenteric neurons [Citation55]. Gain-of-function of the SCN11A gene that encodes NaV1.9 is in most cases associated with pain syndromes and SFN [Citation52,Citation59].
Voltage-gated potassium channels
Voltage-gated potassium channels (VGKCs) are like VGSCs expressed on all neurons. They display a similar membrane topology as the NaV-channel and form complexes with other membrane-bound proteins. Their main responsibility is to repolarize the membrane potential and maintain the cell membrane resting potential. Dysfunction of the VGKC is associated with reduced repolarization, which keeps the cell membrane in a hyperexcitable state [Citation60]. Dysfunction of VGKCs and various proteins that form complexes with VGKC in the human peripheral nervous system are associated with peripheral nerve hyperexcitability (PNH) disorders, that can be inherited or acquired [Citation60]. In PNH, autonomic, motor, and sensory nerves are hyperexcitable and display spontaneous firing properties [Citation40,Citation60].
Acquired, diffuse PNH can be immune-mediated and caused by autoimmune syndromes, para-neoplastic phenomenon, toxins, drugs, and infections, but also idiopathic PNH exists. Contactin-associated protein-2 (Caspr2) and contactin-2 (CNTN2) are proteins found in the juxtaparanodes of peripheral myelinated axons that form complexes with VGKC. In immune-mediated PNH, autoantibodies against the VGKC complexes have been detected [Citation40,Citation60]. Sera from PNH patients added to cultured DRG neurons caused hyperexcitability, and the potassium currents were reduced. The PNH patients were positive for VGKC complex antibodies, suggesting that the antibodies cause the disease by disrupting the repolarizing properties of VGKCs [Citation40,Citation60].
Other mechanisms
Whereas lactate is an oxidative energy source for neurons, enhanced lactate accumulation leads to acidosis, which may lead to neuronal swelling and death. Lactate exerts its effects on cells that have acid-sensing ion channels. In diabetes, hyperglycemia may increase the lactate accumulation [Citation19].
Sterile alpha and toll-like interleukin 1 receptor (TIR) motif containing 1 (SARM1) is a multidomain protein which consists of an autoinhibitory N-terminal domain, tandem SARM1 domains that mediate multimerization, and a C-terminal TIR domain [Citation61]. In healthy axons, SARM1 is predominantly in the off state [Citation62]. Upon generating SARM1 knockout human pluripotent stem cell lines, the metabolic and prodegenerative role of SARM1 was observed in human neurons. The loss of SARM1 blocked the axonal degeneration in human sensory neurons both after traumatic injury and vincristine-induced peripheral neuropathy. These findings suggest a role for SARM1 to prevent and treat human degenerative diseases [Citation63].
A genome-wide association study (GWAS) revealed a low frequency of a loss-of-function splice donor variant in peripherin (PRPH) that was associated with both decreased neural conduction amplitude and an increased risk of an early-onset axonal polyneuropathy in homozygous carriers. From a causal point of view, PRPH encodes for peripherin, a class III intermediate filament (IF) protein which is involved in the cytoskeleton function of the axon [Citation64].
Allodynia and other forms of abnormal pain responses in diffuse peripheral neuropathy
Nociception is afferently conveyed mainly via thin myelinated Aδ fibers and unmyelinated C fibers [Citation65]. Patients with traumatic peripheral nerve injury as well as patients with central lesions may develop peripheral neuropathic pain, which may also be manifested as so-called dynamic mechanical allodynia (DMA). DMA means a pain evoked by a normally non-painful, light mechanical stimulus on the skin. In contrast to normal nociceptive signals, DMA is mediated mainly via type Aβ fibers, both in subjects with peripheral and central pain [Citation66]. In similarity to inflammatory neuropathies, NO influences neuropathic pain. Moreover, overactivation of the N-methyl-D-aspartate receptor (NMDA) receptor leads to increased levels of neuronal and endothelial NO synthase, implying that these enzymes and their products seem to be important in peripheral pain hypersensitivity [Citation67]. Tetrahydrobiopterin (BH4) is a co-factor in the synthesis of NO, serotonin, dopamine, epinephrine, and norepinephrine. Animal studies have shown that the levels of BH4 rise following damage to peripheral nerves and inflammation, in turn promoting a few pain-exaggerating changes, including the activation of the phosphatidylinositol-3 kinases pathway which is involved in the hypersensitivity of neuropathic pain. Human genetic studies have found that patients with a specific haplotype leading to less activation of the rate-limiting enzyme for BH4 production, GTP cyclohydrolase 1 (GCH1), experience less pain in various settings [Citation68]. This finding supports that BH4 is relevant also in human neuropathic pain. Central mechanisms are of importance also in diabetic neuropathy and other forms of peripheral neuropathy [Citation19].
Pain is a common clinical problem in Parkinson’s disease. Even though central mechanisms are thought to be important, pain in Parkinson’s disease likely also stems from damage of peripheral autonomic and somatosensory neurons. It has been suggested that the sensory neuropathy in Parkinson’s disease may partly be caused by lipid alterations, specifically the accumulation of glucosylceramides in plasma [Citation69].
Increased sympathetic activity has been found to be associated with several conditions leading to abnormal pain, e.g., diabetic neuropathy. More specifically, increased peripheral alpha1-receptor activity have been implicated in these pain states [Citation70]. No single study was identified which could explain why some patients develop mainly large fiber neuropathy and other mainly SFN.
Discussion
Several mechanisms may lead to peripheral neuropathy. The most studied mechanism is the pathophysiology behind diabetic neuropathy. However, these cellular mechanisms may be applied also to other disease states. As an example, mitochondrial dysfunction is central in diabetic neuropathy. Several forms of genetic mitochondrial disorders share similar mechanisms behind neuropathy and GI dysmotility [Citation71,Citation72]. Inflammatory mechanisms are observed also in neuropathy secondary to systemic autoimmune diseases with perivascular inflammation and ischemia [Citation6,Citation7]. The loss of myenteric neurons in mice after a Western diet seemed to be mediated by the increased plasma levels of free fatty acids [Citation73], as also are involved in diabetic neuropathy [Citation18].
The most important is to recognize enteric neuropathy, which in many cases render weak and diffuse clinical signs. An increased awareness of simultaneous damages throughout the peripheral nervous system, and less invasive examinations, would improve the care of patients with GI dysmotility. Through a careful anamnesis, comorbidities and exposure to infectious or other agents may be identified, and relatives with similar symptoms can be reported. Wide indications for control of hyperglycemia must be applied, since impaired glucose homeostasis may induce neuropathy before manifest diabetes [Citation20]. To screen for polyneuropathy, temperature or pinprick sensation (small fiber function) and vibration sensation (large fiber function) tests should be performed [Citation3]. Screening with a variety of autoantibodies may be considered; also, antibodies not analyzed at routine controls should be considered [Citation37,Citation43]. In several conditions, malnutrition may be observed secondary to celiac disease, malignancy and so on. Malnutrition is a well-known etiology to neuropathy and not considered in this review. However, malnutrition may occur in several subjects in the general population due to inadequate dietary habits [Citation74], an observation often overlooked in the Western world with an abundance of food. Wide-spread use of proton pumps inhibitors may lead to malabsorption of micronutrients [Citation75]. Objective identification of the neuropathy is important to differ between peripheral neuropathy, central sensitization syndromes, and psychological factors [Citation68], which render quite different treatment strategies. Gastric emptying tests and various manometry examinations reveal dysmotility, but advanced methods for measuring GI function are only available on a few centers, with difficulties in interpreting the results [Citation2]. On the other hand, cardiovascular autonomic nerve tests, electroneurography, electromyography, and skin biopsies for determination of intra-epidermal small nerve fiber density are available at more hospitals and are associated with small risks for the patients. These examinations should be considered more frequently and possibly lead to a reasonable assumption of damage in the enteric nervous system as well, without having to perform a full-thickness bowel biopsy or antroduodenal manometry [Citation2,Citation3,Citation10,Citation11].
If we are familiar with mechanisms behind neuropathy, this complication may be prevented or postponed. Identification of etiology may be of importance to provide targeted treatments, instead of symptomatic treatments often given to reduce the symptoms [Citation3,Citation10]. The most important step to prevent diabetic neuropathy is to optimize the metabolic control [Citation21]. Even though painful diabetic neuropathy originally stems from the peripheral nervous system, the changes of increased pain sensitivity become imprinted in the central nervous system [Citation19]. It is believed that microglia, which are immune cells of the central nervous system, become activated due to a persistent noxious stimulus in the peripheral tissues. Moreover, hyperglycemia may have a direct effect on the microglia. The persistent activation of the microglia is creating a neuroinflammatory response that activates the sympathetic nervous system and hypothalamic-pituitary complex. This leads to an increased sensation to pain also in the central nervous system, a phenomenon known as central sensitization. In diabetic neuropathy, especially spinal microglia have been of particular interest [Citation19].
Most of our knowledge about channelopathies in humans are derived from genetic studies on rare, often inherited diseases. The properties of the various ion channels, such as VGSCs and VGKCs, have been extensively studied in animal models, but its complex nature with inflammation, enhanced transcription, and increased phosphorylation by various effector kinases of the intracellular N-terminal of the VGSC are difficult to study in humans [Citation40,Citation52]. Indirect evidence points in the same direction as in animals. Elevated levels of NaV channel mRNA, as well as MAP kinases, have been detected in blind nerve endings of resected painful human neuromas [Citation33,Citation52,Citation76], with increased ectopic impulse generation. Nevertheless, channelopathy is seldom or never considered in clinical praxis in patients with GI dysmotility. This entity may be under-diagnosed, due to rare genetic examinations. Genetic testing should be considered, both in sporadic and hereditary cases of severe idiopathic peripheral neuropathy.
Conclusions
In conclusion, several different mechanisms may be involved in the development of diffuse peripheral neuropathy involving both internal organs and skin. A thorough anamnesis about the patient and relatives could be helpful to identify possible mechanisms. Screening for metabolic changes and autoantibodies should be widely used. Genetic analysis of genes encoding for ion channels may be considered in the effort to find possible etiology to idiopathic peripheral neuropathy leading to GI dysmotility among many other symptoms.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Roth B, Schiro DB, Ohlsson B. Diseases which cause generalized peripheral neuropathy: a systematic review. Scand J Gastroenterol. 2021;56(9):1000–1010.
- Kamperidis N, Nightingale J. Neurological disorders and small bowel dysmotility. Curr Opin Gastroenterol. 2022;38(3):299–306.
- Pop-Busui R, Boulton AJ, Feldman EL, et al. Diabetic neuropathy: a position statement by the American diabetes association. Diabetes Care. 2017;40(1):136–154.
- Sasaki H, Kawamura N, Dyck PJ, et al. Spectrum of diabetic neuropathies. Diabetol Int. 2020;11(2):87–96.
- DiBaise JK, Harris LA, Goodman B. Postural tachycardia syndrome (POTS) and the GI tract: a primer for the gastroenterologist. Am J Gastroenterol. 2018;113(10):1458–1467.
- Moore PM, Cupps TR. Neurological complications of vasculitis. Ann Neurol. 1983;14(2):155–167.
- Perzyńska-Mazan J, Maślińska M, Gasik R. Neurological manifestations of primary sjögren’s syndrome. Reumatologia. 2018;56(2):99–105.
- Savica R, Carlin JM, Grossardt BR, et al. Medical records documentation of constipation preceding parkinson disease: a case-control study. Neurology. 2009;73(21):1752–1758.
- Themistocleous AC, Ramirez JD, Serra J, et al. The clinical approach to small fibre neuropathy and painful channelopathy. Pract Neurol. 2014;14(6):368–379.
- Schofield JR. Autonomic neuropathy-in its many guises-as the initial manifestation of the antiphospholipid syndrome. Immunol Res. 2017;65(2):532–542.
- Oaklander AL. Immunotherapy prospects for painful small-fiber sensory neuropathies and ganglionopathies. Neurotherapeutics. 2016;13(1):108–117.
- Levine JD, Alessandri-Haber N. TRP channels: targets for the relief of pain. Biochim Biophys Acta. 2007;1772(8):989–1003.
- Garrison SR, Stucky CL. The dynamic TRPA1 channel: a suitable pharmacological pain target? Curr Pharm Biotechnol. 2011;12(10):1689–1697.
- Moher D, Liberati A, Tetzlaff J, PRISMA Group, et al. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097.
- Liberati A, Altman DG, Tetzlaff J, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ. 2009;339:b2700.
- Tomlinson DR, Gardiner NJ. Diabetic neuropathies: components of etiology. J Peripher Nerv Syst. 2008;13(2):112–121.
- Meldgaard T, Keller J, Olesen AE, et al. Pathophysiology and management of diabetic gastroenteropathy. Therap Adv Gastroenterol. 2019;12:1756284819852047.
- Gonçalves NP, Vaegter CB, Andersen H, et al. Schwann cell interactions with axons and microvessels in diabetic neuropathy. Nat Rev Neurol. 2017;13(3):135–147.
- Rahman MH, Jha MK, Suk K. Evolving insights into the pathophysiology of diabetic neuropathy: implications of malfunctioning glia and discovery of novel therapeutic targets. Curr Pharm Des. 2016;22(6):738–757.
- Papanas N, Vinik AI, Ziegler D. Neuropathy in prediabetes: does the clock start ticking early? Nat Rev Endocrinol. 2011;7(11):682–690.
- Calcutt NA. Diabetic neuropathy and neuropathic pain: a (con)fusion of pathogenic mechanisms? Pain. 2020;161(Suppl 1):S65–s86.
- Chandrasekaran K, Anjaneyulu M, Choi J, et al. Role of mitochondria in diabetic peripheral neuropathy: influencing the NAD(+)-dependent SIRT1-PGC-1α-TFAM pathway. Int Rev Neurobiol. 2019;145:177–209.
- Fernyhough P, McGavock J. Mechanisms of disease: mitochondrial dysfunction in sensory neuropathy and other complications in diabetes. Handb Clin Neurol. 2014;126:353–377.
- Nazıroğlu M, Dikici DM, Dursun S. Role of oxidative stress and Ca2+ signaling on molecular pathways of neuropathic pain in diabetes: focus on TRP channels. Neurochem Res. 2012;37(10):2065–2075.
- Jack M, Wright D. Role of advanced glycation endproducts and glyoxalase I in diabetic peripheral sensory neuropathy. Transl Res. 2012;159(5):355–365.
- Ziegler D, Buchholz S, Sohr C, et al. Oxidative stress predicts progression of peripheral and cardiac autonomic nerve dysfunction over 6 years in diabetic patients. Acta Diabetol. 2015;52(1):65–72.
- Verkhratsky A, Fernyhough P. Mitochondrial malfunction and Ca2+ dyshomeostasis drive neuronal pathology in diabetes. Cell Calcium. 2008;44(1):112–122.
- Alecu I, Tedeschi A, Behler N, et al. Localization of 1-deoxysphingolipids to mitochondria induces mitochondrial dysfunction. J Lipid Res. 2017;58(1):42–59.
- Fujita Y, Murakami T, Nakamura A. Recent advances in biomarkers and regenerative medicine for diabetic neuropathy. Int J Mol Sci. 2021;22(5):2301.
- Kobayashi M, Zochodne DW. Diabetic neuropathy and the sensory neuron: new aspects of pathogenesis and their treatment implications. J Diabetes Investig. 2018;9(6):1239–1254.
- Landowski LM, Dyck PJ, Engelstad J, et al. Axonopathy in peripheral neuropathies: mechanisms and therapeutic approaches for regeneration. J Chem Neuroanat. 2016;76(Pt A):19–27.
- Anand P. Neurotrophic factors and their receptors in human sensory neuropathies. Prog Brain Res. 2004;146:477–492.
- Jensen TS, Finnerup NB. Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. Lancet Neurol. 2014;13(9):924–935.
- Skundric DS, Lisak RP. Role of neuropoietic cytokines in development and progression of diabetic polyneuropathy: from glucose metabolism to neurodegeneration. Exp Diabesity Res. 2003;4(4):303–312.
- Elzinga S, Murdock BJ, Guo K, et al. Toll-like receptors and inflammation in metabolic neuropathy; a role in early versus late disease? Exp Neurol. 2019;320:112967.
- Körei AE, Istenes I, Papanas N, et al. Small-Fiber neuropathy: a diabetic microvascular complication of special clinical, diagnostic, and prognostic importance. Angiology. 2016;67(1):49–57.
- Cortese A, Lombardi R, Briani C, et al. Antibodies to neurofascin, contactin-1, and contactin-associated protein 1 in CIDP: clinical relevance of IgG isotype. Neurol Neuroimmunol Neuroinflamm. 2020;7(1):e639.
- Koike H, Katsuno M. Pathophysiology of chronic inflammatory demyelinating polyneuropathy: insights into classification and therapeutic strategy. Neurol Ther. 2020;9(2):213–227.
- Vernino S. Autoimmune autonomic disorders. Continuum (Minneap Minn). 2020;26(1):44–57.
- Faivre-Sarrailh C, Devaux JJ. Neuro-glial interactions at the nodes of ranvier: implication in health and diseases. Front Cell Neurosci. 2013;7:196.
- Rios JC, Melendez-Vasquez CV, Einheber S, et al. Contactin-associated protein (caspr) and contactin form a complex that is targeted to the paranodal junctions during myelination. J Neurosci. 2000;20(22):8354–8364.
- Charles P, Tait S, Faivre-Sarrailh C, et al. Neurofascin is a glial receptor for the paranodin/caspr-contactin axonal complex at the axoglial junction. Curr Biol. 2002;12(3):217–220.
- Sántha P, Dobos I, Kis G, et al. Role of gangliosides in peripheral pain mechanisms. Int J Mol Sci. 2020;21(3):1005.
- Uncini A. A common mechanism and a new categorization for anti-ganglioside antibody-mediated neuropathies. Exp Neurol. 2012;235(2):513–516.
- Pardo CA, McArthur JC, Griffin JW. HIV neuropathy: insights in the pathology of HIV peripheral nerve disease. J Peripher Nerv Syst. 2001;6(1):21–27.
- Lehmann HC, Köhne A, Meyer zu Hörste G, et al. Role of nitric oxide as mediator of nerve injury in inflammatory neuropathies. J Neuropathol Exp Neurol. 2007;66(4):305–312.
- Asahchop EL, Branton WG, Krishnan A, et al. HIV-associated sensory polyneuropathy and neuronal injury are associated with miRNA-455-3p induction. JCI Insight. 2018;3(23):e122450.
- Galligan JJ. HIV, opiates, and enteric neuron dysfunction. Neurogastroenterol Motil. 2015;27(4):449–454.
- Harboe M, Aseffa A, Leekassa R. Challenges presented by nerve damage in leprosy. Lepr Rev. 2005;76(1):5–13.
- Kumar V, Sachan T, Natrajan M, et al. High resolution structural changes of schwann cell and endothelial cells in peripheral nerves across leprosy spectrum. Ultrastruct Pathol. 2014;38(2):86–92.
- Martinez AR, Nunes MB, Nucci A, et al. Sensory neuronopathy and autoimmune diseases. Autoimmune Dis. 2012;2012:873587.
- Cheng X, Choi JS, Waxman SG, et al. Mini-review - Sodium channels and beyond in peripheral nerve disease: modulation by cytokines and their effector protein kinases. Neurosci Lett. 2021;741:135446.
- Hoeijmakers JG, Merkies IS, Gerrits MM, et al. Genetic aspects of sodium channelopathy in small fiber neuropathy. Clin Genet. 2012;82(4):351–358.
- Tibbs GR, Posson DJ, Goldstein PA. Voltage-Gated ion channels in the PNS: novel therapies for neuropathic pain? Trends Pharmacol Sci. 2016;37(7):522–542.
- Sopacua M, Hoeijmakers JGJ, Merkies ISJ, et al. Small-fiber neuropathy: expanding the clinical pain universe. J Peripher Nerv Syst. 2019;24(1):19–33.
- Blesneac I, Themistocleous AC, Fratter C, et al. Rare NaV1.7 variants associated with painful diabetic peripheral neuropathy. Pain. 2018;159(3):469–480.
- Faber CG, Hoeijmakers JG, Ahn HS, et al. Gain of function Naν1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol. 2012;71(1):26–39.
- Han C, Huang J, Waxman SG. Sodium channel Nav1.8: emerging links to human disease. Neurology. 2016;86(5):473–483.
- Kharatmal SB, Singh JN, Sharma SS. Voltage-Gated sodium channels as therapeutic targets for treatment of painful diabetic neuropathy. Mini Rev Med Chem. 2015;15(14):1134–1147.
- Küçükali CI, Kürtüncü M, Akçay H, et al. Peripheral nerve hyperexcitability syndromes. Rev Neurosci. 2015;26(2):239–251.
- Gerdts J, Summers DW, Sasaki Y, et al. Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J Neurosci. 2013;33(33):13569–13580.
- Sasaki Y, Engber TM, Hughes RO, et al. cADPR is a gene dosage-sensitive biomarker of SARM1 activity in healthy, compromised, and degenerating axons. Exp Neurol. 2020;329:113252.
- Chen YH, Sasaki Y, DiAntonio A, et al. SARM1 is required in human derived sensory neurons for injury-induced and neurotoxic axon degeneration. Exp Neurol. 2021;339:113636.
- Bjornsdottir G, Ivarsdottir EV, Bjarnadottir K, et al. A PRPH splice-donor variant associates with reduced sural nerve amplitude and risk of peripheral neuropathy. Nat Commun. 2019;10(1):1777.
- Tsuda M, Koga K, Chen T, et al. Neuronal and microglial mechanisms for neuropathic pain in the spinal dorsal horn and anterior cingulate cortex. J Neurochem. 2017;141(4):486–498.
- Landerholm ÅH, Hansson PT. Mechanisms of dynamic mechanical allodynia and dysesthesia in patients with peripheral and Central neuropathic pain. Eur J Pain. 2011;15(5):498–503.
- Ahlawat A, Rana A, Goyal N, et al. Potential role of nitric oxide synthase isoforms in pathophysiology of neuropathic pain. Inflammopharmacology. 2014;22(5):269–278.
- Latremoliere A, Costigan M. GCH1, BH4 and pain. Curr Pharm Biotechnol. 2011;12(10):1728–1741.
- Klatt-Schreiner K, Valek L, Kang JS, et al. High glucosylceramides and low anandamide contribute to sensory loss and pain in parkinson’s disease. Mov Disord. 2020;35(10):1822–1833.
- Teasell RW, Arnold JM. Alpha-1 adrenoceptor hyperresponsiveness in three neuropathic pain states: complex regional pain syndrome 1, diabetic peripheral neuropathic pain and Central pain states following spinal cord injury. Pain Res Manag. 2004;9(2):89–97.
- Bates MG, Bourke JP, Giordano C, et al. Cardiac involvement in mitochondrial DNA disease: clinical spectrum, diagnosis, and management. Eur Heart J. 2012;33(24):3023–3033.
- Bianco F, Lattanzio G, Lorenzini L, et al. Novel understanding on genetic mechanisms of enteric neuropathies leading to severe gut dysmotility. Eur J Histochem. 2021;65(s1):3289.
- Reichardt F, Chassaing B, Nezami BG, et al. Western diet induces colonic nitrergic myenteric neuropathy and dysmotility in mice via saturated fatty acid- and lipopolysaccharide-induced TLR4 signalling. J Physiol. 2017;595(5):1831–1846.
- Roth B, Larsson E, Ohlsson B. Poor intake of vitamins and minerals is associated with symptoms among patients with irritable bowel syndrome. J Gastroenterol Hepatol. 2022;37(7):1253–1262.
- Corleto VD, Festa S, Di Giulio E, et al. Proton pump inhibitor therapy and potential long-term harm. Curr Opin Endocrinol Diabetes Obes. 2014;21(1):3–8.
- Attal N, Bouhassira D. Mechanisms of pain in peripheral neuropathy. Acta Neurol Scand Suppl. 1999;173:12–24. discussion 48–52.