
Abstract
The human mass balance of lusutrombopag, an orally bioavailable thrombopoietin (TPO) receptor agonist, was characterised in seven healthy male subjects after a single oral dose of [14C]-lusutrombopag (2 mg, 100 μCi) in solution.
Lusutrombopag was the main component in plasma, accounting for 56% of plasma radioactivity AUC0-∞. In plasma, the half-life of radioactivity (70.7 h) was longer than that of lusutrombopag (25.7 h), suggesting the presence of long circulating metabolites. The main excretion pathway of lusutorombopag was feces, with a radioactivity recovery of approximately 83% within 336 h post-dose. M6 (lusutrombopag-O-propanol or lusutrombopag-O-acetic acid) and M7 (lusutrombopag-O-ethane-1,2-diol) were also identified as main components in feces, accounting for at most 17.9%, and 16.9% of the dose, respectively, and were β-oxidation related metabolites.
Our in vitro metabolism study of lusutrombopag indicated that β-oxidation was a subsequent metabolism of ω-oxidation and CYP4 enzymes, including CYP4A11, were the major isozymes contributing to ω-oxidation.
In conclusion, lusutrombopag is primarily eliminated via ω-oxidation and excreted in the feces, where CYP4 enzymes play an important role.
Introduction
Thrombocytopenia is a clinical condition manifested by a decrease in platelets due to increased platelet destruction and/or inadequate platelet production. It results in an increased risk of clinically significant bleeding such as cerebral or other internal haemorrhage, petechiae, purpura, and mucosal bleeding (e.g. epistaxis, gastrointestinal bleeding, and genitourinary bleeding). Excessive bleeding after surgery is also a common risk associated with thrombocytopenia.
Thrombopoietin (TPO) is a cytokine that stimulates proliferation and differentiation of megakaryocytic progenitor cells from hematopoietic stem cells, as well as megakaryocyte maturation, and regulates platelet production (Lok et al. Citation1994, Szilvassy Citation2006). Since TPO can stimulate thrombocytopoiesis, the agonist of TPO receptor has been developed for the treatment of thrombocytopenia.
Lusutrombopag [(2E)-3-{2,6-Dichloro-4-[(4-{3-[(1S)-1-(hexyloxy)ethyl]-2-methoxyphenyl}-1,3-thiazol-2-yl)carbamoyl]phenyl}-2-methylprop-2-enoic acid] () is an orally bioavailable TPO receptor agonist. Lusutrombopag acts on the transmembrane domain of TPO receptors expressed on megakaryocytes, activating signal transduction pathways, such as the Janus kinase − signal transducer and activator of transcription pathway, and the Ras − p44/42 mitogen-activated protein kinase pathway, thus leading to platelet production.
Figure 1. Chemical structure of [14C]-lusutrombopag, with the site of the 14 C label indicated (*).
![Figure 1. Chemical structure of [14C]-lusutrombopag, with the site of the 14 C label indicated (*).](/cms/asset/2bafa0eb-65b8-4a38-bc3e-0b7767f3d634/ixen_a_1845416_f0001_b.jpg)
Lusutrombopag is approved and commercially available for this indication in Japan, the United States, and the European Union. Although a phase 1 single ascending dose study detected M3 and M5 in human plasma by mass spectrometry (unpublished data), detailed elimination pathways of lusutrombopag were not evaluated.
Therefore, it was important to evaluate its metabolism and elimination pathways in humans, which would also provide useful information regarding the risk of drug-drug interactions.
In this study, the pharmacokinetics, metabolism, and mass balance were investigated after a single oral administration of 2 mg [14C]-lusutrombopag to healthy subjects to characterise excretion routes and metabolic pathway in humans. In addition, this study aimed to identify the isozyme responsible for the metabolism of lusutrombopag by conducting an in vitro study using human liver microsomes, human hepatocytes, and recombinant human cytochrome P450 enzymes.
Material and methods
Test compound and chemicals
[14C]-lusutrombopag () was synthesised by Shionogi & Co., Ltd. (Osaka, Japan) with the specific activity of 1.95 MBq/mg and radiochemical purity of 96%. Reference standard for lusutrombopag and its metabolites were synthesised and obtained from Shionogi & Co., Ltd., Japan: M1 (lusutrombopag acyl glucuronide), M2 (taurine conjugate of lusutrombopag β-oxidated carboxylic acid), M3 (lusutrombopag-5-keto), M4 (lusutrombopag β-oxidated carboxylic acid), and M5 (lusutrombopag-O-deshexyl). Internal standards d13-lusutrombopag, d11-M3, and fluoro-substituted-M5 were also supplied by Shionogi & Co., Ltd. Recombinant human cytochrome P450 (CYP) enzymes (CYP1A2, CYP2C9*1, CYP2C19, CYP2D6*1, CYP3A4, CYP4A11, and CYP4F2) were purchased from BD Biosciences (Franklin Lakes, NJ). Pooled Human liver microsomes and cryopreserved human hepatocytes were purchased from Sekisui XenoTech, LLC (Kansas City, KS). Nicotinamide adenine dinucleotide phosphate (reduced form) (NADPH) was purchased from ORIENTAL YEAST CO., LTD (Tokyo, Japan). All other reagents were of high-performance liquid chromatography (HPLC) or guaranteed.
Study design
This was an open-label, non-randomised, single oral dose study involving seven healthy male subjects, aged 20 to 37 years with a body mass index 19.5–29.9 kg/m2. The clinical part of this study was conducted at Quintiles (Durham, NC). Bioanalysis was conducted by Taylor Technology (Princeton, NJ). Radio analysis and metabolite identification was conducted at Analytical Bio-Chemistry Laboratories, Inc. (Columbia, MO).
The study protocol was reviewed and approved by Midlands Institutional Review Board (Overland Park, KS). This study was conducted in full accordance with the Declaration of Helsinki; Good Clinical Practice: Consolidated Guideline approved by the International Conference on Harmonization (ICH); Good Clinical Practices (GCP) as required by and described in 21 Code of Federal Regulations. Informed consent for all subjects was obtained prior to the conduct of any study-related procedures.
Dosing and sample collection
A single oral dose of 2 mg of [14C]-lusutrombopag (approximately 100 μCi) was administered to subjects in a fasted state by an aqueous solution of 36 mM Sodium dihydrogen phosphate, 75 mM sodium hydroxide, and 9.5% Macrogol 400.
Blood samples were collected at the following time points for pharmacokinetic analysis of lusutrombopag and its metabolites (M3 and M5) in plasma and radioactivity in blood and plasma: pre-dose, 0.5, 1, 2, 3, 4, 5, 6, 8, 10, 12, 24, 48, 72, 96, 120, 144, and 168 h post-dose, and then at 24 h intervals until 14 days post-dose. Additional blood samples for metabolite profiling in plasma were collected at the following time points: pre-dose, 1, 2, 4, 8, 12, 24, 48, 72, and 96 h post-dose. Plasma samples were obtained by centrifugation from blood samples at 2000 × G for 15 min at 5 °C. For total radioactivity analysis and metabolite profiling, urine and feces were collected at the following intervals: pre-dose (within 12 h prior to dosing), 0–6, 6–12, and 12–24 h and then at 24 h intervals up to 14 days post-dose for urine, pre-dose (within 24 h prior to dosing), and at 24 h intervals up to 14 days post-dose for feces. All blood, plasma, urine, and fecal samples were stored at or below −70 °C until analysed.
Measurement of radioactivity
Aliquots of plasma and urine were added to a liquid scintillation counting solution. Fecal samples were diluted in water before homogenisation. Aliquots of fecal homogenates and whole blood were combusted using Model 307 Oxidiser (Packard, UK) and the resultant CO2 trapped in Carbo-Sorb® (Perkin Elmer, Waltham, MA) in combination with Permafluor® Ebscintillation cocktail (Perkin Elmer). The radioactivity of all samples was determined by a standard liquid scintillation counting method using LS6000SC and LS6500 (Beckman, Brea, CA).
Quantification of lusutrombopag and its metabolites in plasma
The liquid chromatography with tandem mass spectrometry (LC/MS/MS) consisted of Agilent 1100 HPLC system (Agilent Technologies, Santa Clara, CA) and API5000 (SCIEX, Framingham, MA) and was used to measure concentrations of lusutrombopag, M3, and M5 in plasma. These analytes consisted of a solid phase extraction of plasma and internal standards using an OASIS MAX SPE plate (Waters, Milford, MA). The analyte samples were separated by an XTerra RP8 column (2.1 × 150 mm, 5 μm; Waters, Milford, MA) at 40 °C. The mobile phases used were 0.1% formic acid (FA) in water: acetonitrile: isopropyl alcohol (7:2:1; v/v/v) (A) and 0.1% FA in acetonitrile: water: isopropyl alcohol (7:2:1; v/v/v) (B). The flow rate was 0.4 mL/min in the following linear gradient conditions: hold at 50% B until 0.1 min; 0.1–3.6 min to 100% B; hold at 100% B until 7 min; and return to initial conditions over 1 min. The MS/MS detection was conducted by monitoring the following precursor → fragment ions pairs of 591 → 489 (lusutrombopag), 605 → 482 (M3), and 507 → 257 (M5) coupled to an API5000 with an electrospray ionization in positive ion mode.
Sample preparation for metabolite profiling
Approximately equal volumes (1.5 mL) of plasma from the seven subjects were pooled for each time point. The pooled samples were vortexed to mix with a 3-fold volume of ethanol (EtOH) and centrifuged at 4400 rpm for 10 min to obtain the supernatant. The pellet was extracted with 3 mL of EtOH and centrifuged in the same way. All the supernatants were combined at each time point and evaporated to dryness under N2 gas stream at 35 °C. The residue was reconstituted with 0.6 mL of Dimethylformamide/EtOH/MilliQ water (1:1:1, v/v/v). The samples were centrifuged 14000 rpm for 15 min at room temperature and the obtained supernatants were injected into the HPLC system. The overall extraction recovery from plasma was 90% to 100% at 1 h through 48 h post-dose and 80% and 70% at 72 h and 96 h post-dose, respectively.
Urine and fecal homogenate samples obtained from each subject were analysed individually. A 40 mL aliquot of the urine samples (0–6, 6–12, 12–24, and 24 h intervals up to 216 h post-dose) was pooled, concentrated under N2 gas stream, extracted (Sep-Pak, C18, 20 cc/5 g; Waters Corporation, Milford, MA) and eluted by 1% FA in methanol (MeOH). The eluate was evaporated to dryness under N2 stream at 35 °C. The residue was dissolved with 500 μL of 0.032% FA in dimethylformamide/EtOH/MilliQ water (1:1:1, v/v/v). The samples were centrifuged 14000 rpm for 15 min at room temperature and the obtained supernatants were injected into the HPLC system. The overall extraction recovery from urine was approximately 75% to 95%.
An approximately 10 g aliquot of all the fecal homogenate samples was pooled. The pooled samples were vortexed with a 30 mL volume of 0.2% FA in water and centrifuged at 4400 rpm for 10 min to obtain the supernatant. The pellet was washed with 10 mL of 0.2% FA in water and centrifuged in the same way as the first extraction. This wash procedure was repeated three more times. All the supernatants were extracted (Sep-Pak, C18, 20 cc/5 g) and eluted by 1% FA in MeOH. All the eluates were combined and evaporated to dryness under N2 stream at 35 °C. The residue was dissolved with 1.5 mL of dimethylformamide and 1.5 mL of EtOH. The samples were centrifuged 14000 rpm for 15 min at room temperature and the obtained supernatants were injected into HPLC system. The overall extraction recovery from feces was approximately 65% to 80%.
Metabolite profiling in plasma, urine and feces
Samples from plasma, urine and feces were analysed using an LC/radiochromatography/MS/MS system. Lusutrombopag and its metabolites were identified based on the HPLC retention time and mass spectra of reference compounds. This system comprised an Agilent 1200 HPLC system (Agilent Technologies, Santa Clara, CA), with an API5000 mass spectrometer and a Radiomatic 150TR Flow Scintillation Analyser (Perkin Elmer, Waltham, MA) serving as detectors.
These analyte samples are separated by a Luna® C18(2) column (4.6 × 250 mm, 5 μm; Phenomenex, Torrance, CA) at 40 °C. The mobile phases used were 0.2% FA in water (A) and acetonitrile (B). The flow rate was 1 mL/min in the following linear gradient conditions: starting with 10% B; 0–15 min to 30% B; 15–20 min to 50% B; 20–50 min to 60% B; 50–65 min to 80% B; 65–70 min to 90% B; hold at 90% B until 85 min; return to initial conditions over 5 min. The post-column flow was split at a ratio of 3:7 between the MS and the Radio-Detector. The MS/MS detection was conducted by monitoring the following precursor → fragment ions pairs of 591/593 → 491/493 (lusutrombopag), 767/769 → 491/493 (M1), 700/702 → 491/493 (M2), 605/607 → 491/493 (M3), 593/595 → 491/493 (M4), 507/509 → 257/259 (M5), 565/567 → 491/493 (M6), and 567/569 → 491/493 (M7), using an API5000 with an electrospray ionization in positive ion mode.
Pharmacokinetic analysis
Pharmacokinetic calculations of lusutrombopag and its metabolites in plasma and total radioactivity in plasma and whole blood were performed using noncompartmental techniques with WinNonlin® Version 5.2 (Certara, L.P., Princeton, NJ). The parameters derived were area under the plasma concentration-time curve from time zero to the time of last measurable concentration or infinity (AUC0–t and AUC0–∞, respectively), maximum observed concentration (Cmax), time to Cmax (tmax), apparent terminal elimination half-life (t1/2), and apparent total clearance (CL/F).
Identification of CYP isoforms involved in the formation of lusutrombopag-6-hydroxy and lusutrombopag β-oxidated carboxylic acid
To identify the enzyme involved in the formation of lusutrombopag-6-hydroxy, metabolic reaction of [14C]-lusutrombopag was conducted using cDNA expressed CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, CYP4A11, and CYP4F2. The reaction mixture consisted of 20 μM [14C]-lusutrombopag, 100 pmol P450/mL each CYP enzyme, 1 mM NADPH, and 100 mM potassium phosphate buffer containing 10 mM magnesium chloride, pH 7.4. After pre-incubation at 37 °C, NADPH was added to initiate the enzyme reaction in duplicate and incubation time required 60 min. The reaction was terminated by adding an equal volume of ice-cold acetonitrile and centrifuging at 20800 × G for 1 min at 4 °C to obtain the supernatant.
To identify the enzyme involved in the formation of lusutrombopag-6-hydroxy, an inhibition study of lusutrombopag-6-hydroxy formation in human liver microsomes was conducted using selective CYP inhibitors. To evaluate the inhibitory effect of each inhibitor, [14C]-lusutrombopag was incubated with human liver microsomes in the presence of the CYP inhibitors. The reaction mixture consisted of 20 μM [14C]-lusutrombopag, 0.2 mg/mL pooled human liver microsomes, and 100 mM potassium phosphate buffer containing 10 mM magnesium chloride, pH 7.4 in the presence of 1 mM NADPH and appropriate CYP inhibitors. CYP inhibitors and their concentrations were as follows: α-Naphthoflavone for CYP1A2, 0.5 µM; sulfaphenazole for CYP2C9, 1 µM; ticlopidine for CYP2C19, 1 µM; quinidine for CYP2D6, 0.5 µM; ketoconazole for CYP3A4, 0.5 µM; and HET0016 for CYP4A11 and CYP4F2, 1 µM. After pre-incubation at 37 °C, NADPH was added to initiate the enzyme reaction in duplicate and incubation time required 60 min. The reaction was terminated by adding an equal volume of ice-cold acetonitrile and centrifuging at 20800 × G for 1 min at 4 °C to obtain the supernatant. Human liver microsomes at concentrations of 0.1 to 0.2 mg/mL and incubation times of 0 to 60 min resulted in a linear formation of lusutrombopag-6-hydroxy. A human liver microsomes concentration of 0.2 mg/mL and incubation time of 60 min were therefore selected for inhibition studies.
The enzyme involved in the formation of lusutrombopag β-oxidated carboxylic acid (M4) was identified in an inhibition study of lusutrombopag-6-hydroxy and M4 formation in human hepatocytes using selective CYP inhibitors. To evaluate the inhibitory effect of each inhibitor, [14C]-lusutrombopag was incubated with human hepatocytes in the presence of selective CYP inhibitors. The reaction mixture consisted of 20 μM [14C]-lusutrombopag and 1 × 106 cells/mL human hepatocyte suspension in Williams’ Medium E containing 0.292 g/L L-glutamine. CYP inhibitors and their concentrations were the same as incubation in human liver microsomes. The reaction mixture was incubated in a CO2 incubator (37 °C, 5% CO2) for 4 h, in duplicate. The reaction was terminated by adding 2-fold volume of ice-cold acetonitrile and centrifuging at 14000 rpm for 1 min at 4 °C to obtain the supernatant. Lusutrombopag at concentrations of 5 to 20 μmol/L and incubation times of 2 to 4 h resulted in a linear formation of lusutrombopag-6-hydroxy and lusutrombopag-β-oxidated carboxylic acid. Therefore, the concentration of lusutrombopag and incubation time for inhibition study using human hepatocytes were set at 20 μmol/L and 4 h.
Radio-HPLC and LC(/MS)n analysis for in vitro study
The supernatants of in vitro metabolic reaction samples were dried up by N2 gas and the residues were dissolved with EtOH/0.1% FA in water (4:1, v/v). These analytical samples were analysed by an HPLC/radiochromatography/multistage MS (MSn) system comprised of an Agilent 1100 HPLC system (Agilent Technologies, Santa Clara, CA), with an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, Waltham, MA) and a 625 TR Flow Scintillation Analyser (Perkin Elmer, Waltham, MA) serving as detectors. An XBridgeTM Shield RP18 column (4.6 × 150 mm, 3.5 µm; Waters Corporation, Milford, MA) was used at 40 °C as the analytical HPLC column. The mobile phases used were 0.1% FA in water (A) and acetonitrile (B). The flow rate was 1 mL/min in the following linear gradient conditions: starting with 50% B; 0–20 min to 70% B; 20–30 min to 90% B; hold at 90% B until 35 min; return to initial conditions over 5 min. The post-column flow was split at a ratio of 1:3 between the MS and the Radio-Detector. Conditions of mass spectrometry were as follows: atmospheric pressure ionisation interface, electrospray ionisation; acquisition, full scans in positive ion mode.
In vitro enzyme kinetic analysis
To determine the Michaelis-Menten kinetic parameters (Km and Vmax), [14C]-lusutrombopag was incubated with pooled human liver microsomes over a range of substrate concentrations (5 to 40 μM). Kinetic parameters were calculated by non-linear regression using the WinNonlin Version 5.0.1.
Results
Safety assessment
A single oral dose of 2 mg of [14C]-lusutrombopag was tolerated in the seven subjects. No subjects were withdrawn from the study and no serious adverse events (AE) were observed. The most frequently reported AE was headache in three subjects, but this was mild and resolved prior to the study completion. There were no clinically important changes in clinical laboratory values, vital signs, ECG parameters, and physical examination data during the study.
Pharmacokinetics and excretion
The concentration-time profiles for radioactivity in plasma and blood were similar. (). The onset of absorption appeared to be immediate, with radioactivity and lusutrombopag concentrations quantifiable at the first (0.5 h) post-dose time point with tmax of 5 h, while plasma radioactivity declined slower than lusutrombopag during the elimination phase 24 to 168 h after drug administration. The mean t1/2 of radioactivity was 70.7 h, which was longer than that of lusutrombopag (25.7 h) (), suggesting the presence of metabolites with longer half-life compared with the parent compound. Plasma concentration ratios of M5/radioactivity at all time points were less than 1% and M3 was not quantifiable in any subjects, suggesting that other metabolites contributed slow elimination of radioactivity.
Figure 2. Mean concentration-time profiles of radioactivity in plasma and whole blood and lusutrombopag and M5 in plasma after a single oral administration of 2 mg of [14C]-lusutrombopag to healthy subjects. Each point represents the mean from seven subjects. All M3 concentrations were below the limit of quantitation.
![Figure 2. Mean concentration-time profiles of radioactivity in plasma and whole blood and lusutrombopag and M5 in plasma after a single oral administration of 2 mg of [14C]-lusutrombopag to healthy subjects. Each point represents the mean from seven subjects. All M3 concentrations were below the limit of quantitation.](/cms/asset/a260e2e8-2886-4b81-8c87-5405b36b9b1f/ixen_a_1845416_f0002_b.jpg)
Table 1. Pharmacokinetic parameters of lusutrombopag and M5 and total radioactivity in plasma and whole blood after a single oral administration of 2 mg of [14C]-lusutromobpag.
In the excretion study, a mean of 84.2% of the administered radioactivity was recovered within 336 h with more than half of the dose (60%) excreted by 120 h, indicating that the main excretion pathway of drug-related components was in the feces.
Metabolite profiling in plasma, urine, and feces
HPLC radiochromatogram showed that lusutrombopag was the most abundant radioactive component in plasma, urine, and feces (, ). Although no metabolites were detected in plasma, 10 and 15 metabolites were detected in urine and feces, respectively. M1 and M5 accounted for 0.02% and 0.03%, respectively, of the dose in urine. M1, M2, M3, M4, and M7 accounted for 1.59%, 0.66%, 2.04%, 1.53%, and 16.9% of the dose in feces, respectively. M5 and M6 were eluted as PF3 at the same time and accounted for 17.9% of the dose in feces. The LC/MS peak intensity of M6 was about eight times as that of M5, suggesting the PF3 was mainly constituted by M6 (data not shown). M1, M2, M3, M4, and M5 were identified by authentic standards and M6 and M7 were structurally estimated by MS/MS fragmentations (). M6 and M7 corresponded to the protonated molecules at m/z 565 and 567, respectively, which were 26 Da and 24 Da smaller than lusutrombopag. The product ion at m/z 491 for M6 and M7 was also observed for lusutrombopag, indicating that metabolic reaction should happen on the O-hexyl chain. The 26 Da or 24 Da loss of molecular weight suggested shortening of the alkyl chain and oxidation at the O-hexyl moiety. Hence, M6 could be an O-propanol or O-acetic acid and M7 could be the O-ethane-1,2-diol. Based on these data, the proposed metabolic pathways of lusutrombopag in humans are illustrated in . As for M2, M4, M6, and M7, the number of carbon atoms at the O-alkyl chain suggested stepwise alkyl shortening reactions from lusutrombopag-6-hydroxy such as seen with fatty acid metabolism. These four metabolites accounted for at most 37.0% of the dose in feces, indicating ω-oxidation predominantly contributed to elimination of lusutrombopag.
Figure 3. Representative HPLC-radiochromatograms of plasma collected from 8 h, urine collected from 0 to 216 h, and feces collected from 0 to 336 h after single oral administration of 2 mg of [14C]-lusutrombopag to healthy subjects.
![Figure 3. Representative HPLC-radiochromatograms of plasma collected from 8 h, urine collected from 0 to 216 h, and feces collected from 0 to 336 h after single oral administration of 2 mg of [14C]-lusutrombopag to healthy subjects.](/cms/asset/2998857c-2f23-4466-adb9-c6646d0b67c9/ixen_a_1845416_f0003_c.jpg)
Figure 4. Postulated metabolic pathways of lusutrombopag in humans.
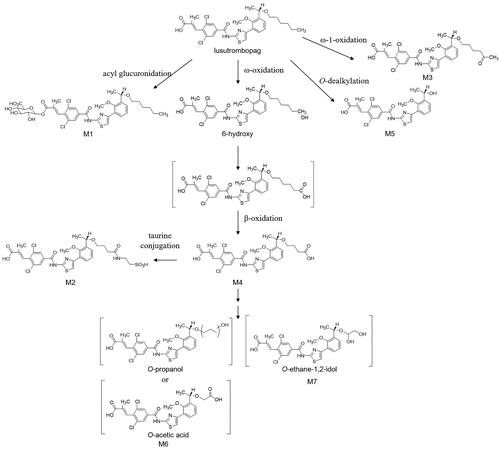
Table 2. Composition of lusutrombopag and its metabolites in urine and feces after a single oral administration of 2 mg of [14C]-lusutromobopag, and assignment of MS/MS fragmentations of unidentified metabolites.
Identification of CYP isozymes involved in the formation of lusutrombopag-6-hydroxy
The formation of lusutrombopag-6-hydroxy was observed by the CYP2C19 (591 fmol/h/pmol P450), CYP3A4 (78.3 fmol/h/pmol P450), and CYP4A11 (143 fmol/h/pmol P450) (). The contribution of other recombinant CYP enzymes was minimal. The formation of lusutrombopag-6-hydroxy was inhibited by the CYP4 inhibitor HET0016 (58.2%), the CYP3A4 inhibitor ketoconazole (46.0%), and the CYP2C19 inhibitor ticlopidine (20.3%) (). The other selective inhibitors inhibited the formation of lusutrombopag-6-hydroxy by approximately 10%. These results suggest that CYP2C19, CYP3A4, and CYP4A11 were the enzymes responsible for the formation of lusutrombopag-6-hydroxy.
Table 3. In vitro metabolism of [14C]-lusutrombopag by recombinant human cDNA expressed CYP enzymes.
Table 4. Effect of P450 chemical inhibitors on the formation of lusutrombopag-6-hydroxy from [14C]-lusutrombopag following incubation with human liver microsomes.
Identification of CYP isozymes contributing to the formation of lusutrombopag-β-oxidated carboxylic acid in human hepatocytes
Lusutrombopag β-oxidated carboxylic acid (M4) was assumed to be produced from lusutrombopag-6-hydroxy in humans. The formation β-oxidated carboxylic acid and lusutrombopag-6-hydroxy was then investigated in human hepatocytes which contained the whole metabolizing enzymes (both phase 1 and 2) in the presence of CYP selective inhibitors (). The CYP4 inhibitor, HET0016, inhibited the formation of lusutrombopag-6-hydroxy (77.2%) and M4 (57.9%). The CYP3A4 inhibitor, ketoconazole, also inhibited the formation of lusutrombopag-6-hydroxy (15.7%) and M4 (24.2%). The other selective inhibitors inhibited the formation of lusutrombopag-6-hydroxy and M4 by less than 10%. CYP4 contributed to the formation of M4 and lusutrombopag-6-hydroxy more than CYP3A4. Ketoconazole and HET0016 inhibited the production of lusutrombopag-6-hydroxy and M4 to the same extent, suggesting that β-oxidation was a subsequent reaction from lusutrombopag-6-hydroxy.
Table 5. Effect of P450 chemical inhibitors on the formation of lusutrombopag-6-hydroxy and lusutrombopa-β-oxidated carboxylic acid from [14C]-lusutrombopag following incubation with cryopreserved human hepatocytes.
Discussion
In this study, we determined the elimination pathway of lusutrombopag and identified the enzyme involved in its metabolism in humans. Following oral administration of [14C]-lusutrombopag solution, the concentration ratios of lusutrombopag/total radioactivity in plasma were 80% at tmax, suggesting most of radioactivity in plasma was unchanged lusutrombopag. However, a lower contribution of lusutrombopag to total exposure was observed with approximately 56% AUC0-∞ ratios of lusutrombopag to radioactivity in plasma. At later time points from 24 h, the decline in plasma radioactivity concentrations was slower than that in lusutrombopag and the contributions of other analyte(s) became more apparent, ranging from 40.2% at 48 h to 85.8% at 168 h. The half-lives of radioactivity, 70.7 h in plasma and 111 h in blood, were longer than that of lusutrombopag in plasma (25.7 h). In addition, metabolite profiling in plasma showed some metabolites accounting for <1% of the dose. It is assumed that multiple metabolites were not detected by radio detector due to the low dose of radio activity (52 kBq/kg). These data suggest the presence of many minor circulating metabolites whose elimination appeared to be slower than that of lusutrombopag.
In blood, the ratios of radioactivity concentrations in whole blood to plasma across all time points were 52.9% to 56.9%. These ratios were comparable to the ratio of volume in plasma to whole blood (approximately 55%) (Seeley et al. Citation2000). These results indicate that [14C]-lusutrombopag was not distributed to blood cells.
The total recovery of radioactivity up to 336 h after dosing was approximately 84% of the dose administered. The excretion rates of radioactivity in feces and urine were approximately 83% and 1% of the dose administered, respectively. A small proportion of total radioactivity was excreted into the feces on the last sampling day, suggesting further recovery of radioactivity from the feces after Day 15. These findings demonstrate that lusutrombopag is excreted mainly via the fecal route with minimal contribution of urinary excretion.
In feces, M6 (including M5) and M7 accounted for at 17.9% and 16.9% of the administrated radioactivity, respectively, with minor contributions of other metabolites (M1, M2, M3 and M4) (<3% of the dose for each). The structures of M2, M4, M6, and M7 lack two to four carbon atoms in the hexyl chain of lusutrombopag, which suggests that there are sequential metabolic reactions consisting of ω-oxidation and β-oxidation. Omega-oxidation is the initial reaction that hydroxylates the terminal carbon atom of the hexyl chain and β-oxidation is the catabolic process by which the alkyl chain is broken down. Therefore, more than 30% of the dose was assumed to be metabolised via ω- and β-oxidation. The same series of reactions is seen with fatty acid metabolism in some species, including humans (Houten and Wanders Citation2010). Since β-oxidation is facilitated by mitochondria or peroxisomes, liver microsomes could not produce β-oxidation-related metabolites. Therefore, human hepatocytes, which include all organelles, are more appropriate to investigate the metabolic profile of the drugs that form β-oxidation-related metabolites such as lusutrombopag.
The responsible metabolizing enzymes for ω-oxidation of lusutrombopag, which was speculated to be a precursor of β-oxidation-related metabolites and the initial step of major metabolic pathway of lusutrombopag, were assessed with recombinant human cytochrome P450 (CYP) enzymes, human liver microsomes, and human hepatocytes with typical CYP inhibitors. The in vitro study using recombinant CYP and human liver microsomes with chemical inhibitors indicated that CYP2C19, CYP3A4, and CYP4A11 was responsible for the formation of lusutrombopag-6-hydroxy. Additionally, the CYP4 inhibitor inhibited the formation of lusutrombopag-6-hydroxy (77.2% inhibition) and M4 (57.9% inhibition) in human hepatocytes. Because the inhibition ratios were similar, lusutrombopag-6-hydroxy would be a precursor of metabolites with β-oxidation. The result showed that the formation of lusutrombopag-6-hydroxy was catalyzed by CYP4 enzymes, including CYP4A11. In addition, the CYP3A4 inhibitor inhibited the formation of lusutrombopag-6-hydroxy (15.7% inhibition), and no inhibition was observed by selective inhibitors of other CYP enzymes. These results suggest that CYP3A4 partially contributed to the formation of lusutrombopag-6-hydroxy. Possible metabolic pathways of lusutrombopag based on in vitro and/or in vivo metabolites found in humans are shown in .
If the enzyme is responsible for ≥25% of drug elimination, guidelines from the European Medicines Agency and the Food and Drug Administration recommend that clinical drug-drug interaction studies should be conducted using strong inhibitors and/or inducers of the enzyme (European Medicines Agency Citation2012, US Food and Drug Administration Citation2017). The β-oxidation-related metabolites of lusutrombopag were recovered up to 37% of the administrated dose. Based on the University of Washington Drug Interaction Database (DIDB), there has been no report that any drugs inhibit or induce CYP4 to a clinically meaningful extent (http://www.druginteractioninfo.org), indicating a low risk of clinical drug interaction as a CYP4 substrate. When the contribution ratio to lusutrombopag metabolism for CYP4 is 37%, CYP4 inhibitor increases the AUC of lusutrombopag at most 1.6-fold (Rodrigues Citation2008). The clinical DDI study with cyclosporine, a CYP3A4 inhibitor, indicated the fold increase of lusutrombopag AUC was 1.19 (Katsube et al. Citation2020), suggesting the contribution of CYP3A4 for this drug metabolism is low. Considering that lusutrombopag was found to be metabolized primarily by CYP4A with minor contributions of CYP3A4 in this study, it could be concluded that the pharmacokinetics of lusutrombopag would be minimally or modestly changed via inhibition or induction for metabolic enzymes.
It is well known that the CYP4 family catalyzes the ω-hydroxylation of fatty acids, including eicosanoids, prostaglandins, leukotrienes, and arachidonic acids (Hardwick Citation2008). On the other hand, there are only a few drugs for which the CYP4 family contributes to their metabolism (Jin et al. Citation2011, Yamane et al. Citation2015). However, a recent study demonstrated that CYP4F enzymes constituted the third largest piece of the human cytochrome P450 (Michaels and Wang Citation2014). To elucidate responsible drug enzymes, drug metabolism researchers should pay attention to the CYP4 family in addition to major CYP isozymes.
In summary, the results of this study clarified the human pharmacokinetics, mass balance, and metabolite profile of lusutrombopag after a single oral administration of 2 mg of [14C]-lusutrombopag in solution. Lusutrombopag accounted for the majority of the systemic radioactivity. Of the administrated dose, 83.1% was excreted in feces. The main metabolites in feces were β-oxidation-related metabolites, lusutrombopag-O-propanol or lusutrombopag-O-acetic acid (M6), and lusutrombopag-O-ethane-1,2-diol (M7), which were considered to be formed from lusutrombopag-6-hydroxy. The isozymes responsible for the formation of lusutrombopag-6-hydroxy were mainly in the CYP4 family, including CYP4A11 and partially CYP3A4. Therefore, lusutrombopag is primarily eliminated in feces via metabolism, where CYP4 family plays an important role.
Disclosure statement
All authors are employees of Shionogi & Co, Ltd. The authors declare no other conflicts of interests.
Additional information
Funding
References
- European Medicines Agency., 2012. Guideline on the investigation of drug interactions [online]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf [Accessed 22 Oct 2020].
- Hardwick, J.P., 2008. Cytochrome P450 omega hydroxylase (CYP4) function in fatty acid metabolism and metabolic diseases. Biochemical pharmacology, 75, 2263–2275.
- Houten, S.M. and Wanders, R.J., 2010. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. Journal of inherited metabolic disease, 33, 469–477.
- Jin, Y., et al., 2011. CYP4F enzymes are responsible for the elimination of fingolimod (FTY720), a novel treatment of relapsing multiple sclerosis. Drug metabolism and disposition, 39, 191–198.
- Katsube, T., et al., 2020. Evaluation of drug-drug interaction of lusutrombopag, a thrombopoietin receptor agonist, via metabolic enzymes and transporters. European journal of clinical pharmacology. Available from: https://link.springer.com/article/10.1007/s00228-020-02960-7#article-info [Accessed 22 October 2020].
- Lok, S., et al., 1994. Cloning and expression of murine thrombopoietin cDNA and stimulation of platelet production in vivo. Nature, 369, 565–568.
- Michaels, S. and Wang, M.Z., 2014. The revised human liver cytochrome P450 "Pie": absolute protein quantification of CYP4F and CYP3A enzymes using targeted quantitative proteomics. Drug metabolism and disposition, 42, 1241–51.
- Rodrigues, A.D., 2008. Drug-drug interactions. 2nd ed. New York, NY: Informa Healthcare.
- Seeley, R.R., Stephens, T.D., and Tate, P., 2000. Anatomy and physiology. New York, NY: McGraw Hill.
- Szilvassy, S.J., 2006. Haematopoietic stem and progenitor cell-targeted therapies for thrombocytopenia. Expert Opinion on Biological Therapy, 6, 983–992.
- US Food and Drug Administration., 2017. In vitro metabolism and transporter mediated drug-drug interaction studies guidance for industry. Available from: https://www.fda.gov/media/108130/download [Accessed 22 Oct 2020].
- Yamane, M., et al., 2015. In vitro profiling of the metabolism and drug-drug interaction of tofogliflozin, a potent and highly specific sodium-glucose co-transporter 2 inhibitor, using human liver microsomes, human hepatocytes, and recombinant human CYP. Xenobiotica, 45, 230–238.