
Abstract
Emvododstat is a potent inhibitor of dihydroorotate dehydrogenase and is now in clinical development for the treatment of acute myeloid leukaemia and COVID-19.
Following an oral dose administration in Long-Evans rats, 14C-emvododstat-derived radioactivity was widely distributed throughout the body, with the highest distribution in the endocrine, fatty, and secretory tissues and the lowest in central nervous system.
Following a single oral dose of 14C-emvododstat in rats, 54.7% of the dose was recovered in faeces while less than 0.4% of dose was recovered in urine 7 days post-dose. Emvododstat was the dominant radioactive component in plasma and faeces.
Following a single oral dose of 14C-emvododstat in dogs, 75.2% of the dose was recovered in faeces while 0.5% of dose was recovered in urine 8 days post-dose. Emvododstat was the dominant radioactive component in faeces, while emvododstat and its two metabolites (O-desmethyl emvododstat and emvododstat amide bond hydrolysis product) were the major circulating radioactivity in dog plasma.
Introduction
Emvododstat, also known as PTC299 (), is an orally bioavailable small molecule drug candidate under clinical development for the treatment of acute myeloid leukaemia (AML) and COVID-19 (coronavirus SARS-CoV-2). Emvododstat was originally identified as an inhibitor of the translation of Vascular Endothelial Growth Factor A (VEGFA) mRNA (Cao et al. Citation2016) and was developed as an oncology agent for the treatment of solid tumours (Packer et al. Citation2015; Ignacio et al. Citation2016; Weetall et al. Citation2016). Later research demonstrated that emvododstat’s mechanism of action is due to its direct and potent inhibition of dihydroorotate dehydrogenase (DHODH) enzyme, a rate-limiting enzyme in de novo pyrimidine nucleotide synthesis (Cao et al. Citation2019; Branstrom et al. Citation2022). In vitro studies showed that emvododstat is more potent against leukaemic malignancies, including AML, than against solid tumours (Cao et al. Citation2019). Emvododstat has also shown broad-spectrum antiviral activity, and in particular, emvododstat potently inhibits viral replication and suppresses induction of inflammatory cytokines in SARS-CoV-2 cell-based assays (Luban et al. Citation2021). Based on these results, emvododstat has the potential to address unmet needs in certain cancers and RNA virus infection diseases where cancer cells or viruses rely on the de novo biosynthesis of pyrimidine nucleotides for survival or rapid proliferation.
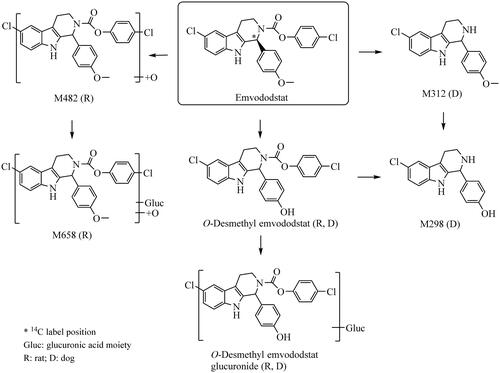
Figure 1. Structure of emvododstat and metabolic pathways of emvododstat in rats and dogs following a single oral dose.
In support of emvododstat clinical development, extensive in vitro and in vivo studies have been conducted to characterise absorption, distribution, metabolism, excretion, and pharmacokinetic drug interaction properties of emvododstat. In vitro metabolism studies showed that O-demethylation followed by glucuronidation is the major metabolic pathways for emvododstat; multiple CYPs appear to be involved in emvododstat metabolism; and emvododstat and O-desmethyl emvododstat are both inhibitors of CYP2D6 and BCRP transporter, but neither of them is a substrate for the common efflux or uptake transporters that were investigated (Ma et al. Citation2022). Following a single oral dose, emvododstat is bioavailable in mice, rats, dogs, and monkeys; the absorption was generally slow, and the plasma exposure of O-desmethyl emvododstat was lower in rodents, but relatively higher in dogs and monkeys (Ma et al. Citation2022). Emvododstat has been administered to healthy subjects (Weetall et al. Citation2016), patients with solid tumours (Packer et al. Citation2015; Ignacio et al. Citation2016) or AML, and now is under clinical development for AML and hospitalised subjects with COVID-19 infection.
The objectives of this study were to evaluate absorption, distribution, metabolism, and excretion (ADME) of emvododstat in rats and dogs following a single oral dose of 14C-emvododstat. Rats and dogs were used for such studies because these species were also used for emvododstat toxicology evaluation. Studies using live animals are essential for ADME property evaluation and are required by regulatory agencies worldwide for investigational drugs. These animal models provide important information for the extrapolation of DMPK and toxicological effects detected in animals to humans. No acceptable non-live animal models are available. The number of animals used in these studies is the minimum required to obtain scientifically and statistically meaningful results. The results from these studies are essential prior to conducting a clinical study to understand human metabolism and excretion of this novel DHODH inhibitor for the potential treatment of COVID-19 infection.
Materials and methods
Materials
14C-Emvododstat (124 µCi/mg, radiochemical purity and chemical purity >99%) was prepared by Pharmaron (Cardiff, UK). Non-labelled emvododstat (chemical purity >99%) and O-desmethyl emvododstat (>97% purity) were synthesised by Siegfried AG (Zofingen, Switzerland).
Methods
ADME studies in laboratory animals
All animal study protocols and procedures employed were ethically reviewed and approved by PTC Therapeutics, Inc. All animal facilities used were fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International, and all animal studies were conducted after approval by the Institutional Animal Care and Use Committee and were in compliance with the US National Research Council’s Guide for the Care and Use of Laboratory Animals, the US Public Health Service’s Policy on Humane Care and Use of Laboratory Animals, and the Guide for the Care and Use of Laboratory Animals. All study protocols (including objectives, study design, and analysis plan) were prepared and approved before the study conducts.
Tissue distribution study in long-evans rats following a single oral dose of 14C-emvododstat
To evaluate potential binding of emvododstat and its metabolites to melanin containing tissues, partially pigmented Long-Evans rats that contain high levels of melanin in skin and uveal tract were used. Eleven intact male Long-Evans rats, approximately 7 weeks old, 169 to 204 g of body weight, were used in this study. Each animal was uniquely identified by a metal ear tag displaying a permanent animal number with the corresponding number written on their tails. All animals were acclimated for approximately 5 days. During the acclimation period, the animals were housed ≤5 per cage in polycarbonate shoebox cage containing hardwood shavings in an environmentally monitored room. A cage card was affixed to each cage displaying the permanent animal numbers. One day prior to and during the in-life testing period animals were housed individually in polycarbonate shoebox cages with stainless steel wire flooring inserts. Animals received Certified Rodent Diet #5002 (PMI Feeds; Brentwood, MO) ad libitum. Fresh tap water was available ad libitum via bottles attached to the cages throughout the study. Environmental conditions such as temperature, humidity, and lighting etc. for the animal room were monitored, and the 12 h dark cycle was maintained at extent possible throughout the study. The animals were examined daily for any signs of abnormalities indicative of health problems. Only animals observed to be healthy were placed on the study. On the day of dosing, each animal was administered a single oral gavage dose of 14C-emvododstat at 50 mg/kg (50 μCi/kg). At 1, 2, 4, 8, 24, 48, 96, 168 and 672 h post-dose, one animal per time point was anaesthetised by inhalation of isoflurane, followed by blood collection via cardiac puncture and euthanasia by isoflurane overdose. Each carcase was then frozen in a dry-ice/hexane bath. Blood and plasma samples were assayed for total radioactivity (TRA) concentrations by liquid scintillation counting (LSC). The tissue concentrations of TRA were determined using quantitative whole-body autoradiography (QWBA) techniques. Briefly, sagittal 30 µm thick sections of the carboxymethylcellulose (CMC)-embedded rat carcases, including quality control (QC) standards, and the CMC-embedded calibration standards, were sectioned using a Leica 9800 Cryomicrotome at −20 °C. The sections were dehydrated for at least 48 h in the cryomicrotome chamber at approximately −20 °C and then transferred to a desiccant storage box for at least 1 h. The sections were removed from their frames, mounted on a support cardboard backing, and labelled with radioactive ink, and the sections were wrapped with plastic wrap. Whole-body sections, containing their QC standards, were placed in phosphor imaging plate (IP) cassettes along with a section containing blood calibration standards. The IPs were exposed to the sections for approximately 4 days at room temperature in a copper-lined lead-shielded box. Autoradioluminograms were generated in a darkroom using a GE Typhoon FLA 9500 Phosphor Imager and analysed using AIDA software. The radioactivity concentrations in selected tissues were determined by digital analysis of the photo-stimulated light/unit area on each autoradioluminogram. A calibration curve was established for each IP and the concentrations of radioactivity in tissues were back calculated and expressed in µg eq/g tissue.
Absorption, metabolism, and excretion (AME) in rats following a single oral dose of 14C-emvododstat
Intact male Sprague Dawley (SD) rats, and bile-duct cannulated (BDC) male SD rats, approximately 7 weeks old, 223 to 257 g of body weight, were used in this study. Each animal was uniquely identified by a permanent tail tattoo number. All animals were acclimated for five days. The intact animals were housed ≤5 per cage in solid bottom polycarbonate holding cages with bedding. BDC rats were individually housed in solid bottom polycarbonate holding cages with bedding. Animals were placed in the testing cages one day prior to dosing and during the in-life testing period. Intact rats for mass balance were housed in individual stainless steel metabolism cages equipped for the separate collection of urine and faeces. BDC rats were housed individually in Nalgene-type metabolism cages, equipped for the separate collection of urine, faeces, and bile. Animals for blood/plasma PK were housed ≤2 per cage in stainless steel wire mesh bottom holding cages. All animals received Certified Rodent Diet #5002 (PMI Nutrition International, Inc.; St. Louis, MO) ad libitum. On the day prior to dose administration, animals were fasted with food returned 4 h post dose. Fresh tap water was available ad libitum via bottles attached to the cages throughout the study. Environmental conditions such as temperature, humidity, and lighting etc. for the animal room were monitored, and the 12 h dark cycle was maintained at extent possible throughout the study. The animals were examined daily for any signs of abnormalities indicative of health problems. Only animals observed to be healthy were placed on the study. On the day of dosing, each administered a single oral gavage dose of 14C-emvododstat at 40 mg/kg (100 μCi/kg). For mass balance in intact rats (n = 3), urine and faeces were collected at 0–8, 8–24 (0–24 for faeces), and every 24 h up to 168 h post-dose. For mass balance in BDC rats (n = 3), urine and faeces were collected at 0–8, 8–24 (0–24 for faeces), 24–48, and 48–72 h post-dose, and bile was collected at 0–4, 4–8, 8–24, 24–48, and 48–72 h post-dose. Bile, urine, and faecal samples were stored at approximately −70 °C before analysis. For plasma pharmacokinetics in intact rats, blood samples were collected at 0, 0.5, 1, 2, 4, 8, 24, 48, 72, 120, and 168 h post-dose (n = 3/timepoint) into syringes containing K2EDTA. Blood samples were centrifuged at approximately 4 °C for 10 min at 1372 g to isolate plasma. All samples were stored at approximately −70 °C before analysis.
AME in dogs following a single oral dose of 14C-emvododstat
Three naïve male beagle dogs, 0.5 to 1 year of age and 6.85 to 7.50 kg of body weight, were used in this study. Each dog was identified by distinct ear tattoos. Dogs were acclimated for at least 7 days and were individually housed in the stainless-steel cages. The animals were examined daily for any signs of abnormalities indicative of health problems. Only healthy dogs were placed on the study. One day prior to and during the in-life testing period, the dogs were housed individually in the stainless-steel metabolism cages equipped for the separate collection of urine and faeces. Environmental conditions such as temperature, humidity, and lighting etc. for the animal room were monitored, and the 12 h dark cycle was maintained at extent possible throughout the study. Dogs received Certified Canine Diet (Certified Dog Diet #20190408, Xietong Inc., Jiangsu, China) ad libitum. All dogs were fasted overnight prior to dosing and for 4 h after dosing. Dogs received drinking water ad libitum. The animals were examined daily for any signs of abnormalities indicative of health problems. Only dogs observed to be healthy were placed on the study. On the day of dosing, animals were each administered a single oral dose of 14C-emvododstat at 20 mg/kg (25 μCi/kg). Blood samples were collected from the cephalic vein into K2EDTA tubes at 0, 1, 2, 4, 8, 12, 24, 48, 72, 120, and 168 h post-dose. Blood samples were centrifuged at 3000 g and 4 °C for 15 min to obtain plasma. Blood samples were stored at −20 °C and plasma samples were stored at −70 °C until analysis. Urine, faeces, cage rinse, and cage wash were collected at 0–8, 8–24 (0–24 for faeces), and every 24 h up to 192 h post-dose. All urine, faeces, and cage rinse samples were stored at −20 °C until analysis.
Following oral dose administration, animals were observed for general health and mortality twice a day (AM and PM) during workdays, and once a day during weekends and holidays. At the completion of the study, the dogs were heathy and returned to the colony.
Sample preparation for TRA analysis
Aliquots of plasma, urine, bile, and cage rise/wash samples from rats and dogs were taken for direct LSC. Rat and dog faecal samples were homogenised with isopropyl alcohol/purified water (1:1, w:w); aliquots of faecal homogenates or blood samples were taken and air-dried at room temperature and combusted in a biological sample oxidiser followed by direct LSC. Carcases from intact rats used for mass balance study were digested in a 6 normal potassium hydroxide solution for approximately 7 days. Aliquots of the carcase digest were taken directly for LSC.
Sample preparation for metabolite profiling and identification
Rat AME study samples
Plasma samples at 2, 4, 8, 24, 48, 72, 120, and 168 h were selected for radio-profiling. An equal volume (approximately 0.5 mL) of each selected plasma sample was pooled by time-point across animals. The pooled plasma samples were extracted with acetonitrile (3×, v/v) for 3 times. The acetonitrile extracts were combined and evaporated to dryness under the nitrogen stream. The residues were reconstituted in acetonitrile: water (70:30) (v/v) for radio-profiling. An equal percentage by volume of individual bile samples at 0–24 h and 24–72 h from male BDC rats were pooled by time interval across rats. A portion of the pooled bile sample was centrifuged at approximately 10,000 g at 4 °C for 10 min. The supernatants were used for radio-profiling. An equal percentage by volume of individual urine samples at 0–24 h and 24–144 h from intact rats and 0–24 h and 24–72 h from BDC rats was pooled by group and time interval across animals, respectively. A portion of the pooled urine sample was centrifuged at 10,000 g at 4 °C for 10 min. The supernatants were evaporated under the nitrogen stream to dryness. The residues were reconstituted with methanol: water (1:1) (v/v) for radio-profiling. An equal percentage by weight of individual faecal samples at 0–24 h and 24–144 h from male intact rats, and at 0–48 h and 48–72 h from BDC rats was pooled by group and time interval across animals, respectively. An aliquot of the pooled faecal homogenate was extracted three times with acetonitrile (3×, v/w). The acetonitrile extracts were combined and evaporated to dryness under the nitrogen stream. The residues were reconstituted with acetonitrile: water (70:30) (v/v) for radio-profiling.
Dog AME study samples
Plasma from each animal at 1, 2, 4, 8, 12, 24, 48, and 72 h were selected and pooled with the same volume by the time points across animals. Each pooled plasma was extracted with methanol (3×, v/v) two times. The methanol extracts were combined and evaporated into dryness under nitrogen stream, and the residues were reconstituted with methanol: water (8:2, v/v) for radio-profiling. An equal percentage sample volume of individual urine samples at 0–8, 8–24, and 24–48 h were pooled across time intervals and animals to obtain a pooled 0–48 h urine sample for metabolite radio-profiling. The pooled urine was mixed with methanol (3×, v/w) followed by evaporation to dryness under the nitrogen stream. The residue was reconstituted with methanol: water (8:2, v/v) for radio-profiling. An equal percentage sample weight of individual faecal samples at 0–24, 24–48, 48–72, and 72–96 h were pooled across time intervals and animals, resulting in 3 pooled faecal samples (0–24, 24–48, 0–96 h) for metabolite radio-profiling. Each pooled faecal homogenate was extracted three times with methanol (3×, v/w), the methanol extracts were combined and evaporated to dryness under the nitrogen stream. The residues were reconstituted with methanol: water (8:2, v/v) for radio-profiling.
Metabolite radio-profiling and identification
The metabolite profiles were determined by high performance liquid chromatography (HPLC) radio-chromatography using a Waters 2695 or Shimadzu UFLC CBM-20A system. An ACE 3 C18 AR column, 3 μm, 150 × 4.6 mm, maintained at 30 °C, and two solvent systems of 0.1% formic acid and 2 mM ammonium acetate in water: acetonitrile (95:5, v/v) (A), and 0.1% formic acid and 2 mM ammonium acetate in acetonitrile: water (95:5, v/v) (B) were used. The flow rate was 0.7 mL/min and the linear gradients were: 5% B for 3 min; 5 to 25% B in 2 min; 25 to 50% B in 25 min, 50 to 95% B in 10 min, hold 95% B for 5 min; 95 to 100% B in 5 min and hold 100% B for 3 min; 100 to 5% B in 1 min and hold 5% B for 15 min. HPLC fractions were collected by time (15 sec/fraction) into Deepwell LumaPlateTM-96 plates. The plates were subsequently dried by a SpeedVac concentrator for up to 8 h. The radioactivity in each fraction was determined by Packard TopCount NXTTM Microplate Scintillation and Luminescence Counter technology. HPLC radio-chromatograms were reconstructed and the radioactivity peaks were integrated to determine the percent distribution of individual radioactivity peaks or regions in each sample. Plasma, bile, urine, and faecal metabolites were analysed by LC-MS/MS using a Thermo Q Extractive mass spectrometer (Thermo Fisher Scientific, Waltham, MA, US). The Q Extractive was equipped with an electrospray ionisation source operated in positive or negative ion mode with a capillary temperature of 350 °C and spray voltage of 3.5 kV. The sheath gas, auxiliary gas, and sweep gas pressure was 60, 20, and 10 units, respectively.
Pharmacokinetic analysis
Pharmacokinetic (PK) data were generated by non-compartmental PK analysis using WinNonlin™ (Version 6.1 or higher, Pharsight Corporation; Mountain View, CA, US). The following parameters were calculated to the extent possible: maximum concentration (Cmax), time of Cmax (Tmax), terminal half-life (T1/2), and area under the concentration versus time curve (AUC).
Results
Tissue distribution in rats following a single oral 14C-emvododstat dose
Following a single 50 mg/kg oral dose of 14C-emvododstat in male Long-Evans rats, the dose was well tolerated. The animals appeared to be normal throughout the study and demonstrated no clinical signs from the drug administration.
Following a single 50 mg/kg oral dose of 14C-emvododstat in rats, both blood and plasma TRA peaked at 8 h, decreased steadily after Tmax, and were below the quantifiable limit (BQL) by 672 h post-dose. Most tissues had Tmax values at 8 h post-dose and the concentrations were BQL (0.495 µg eq/g) by 672 h post-dose, except the epididymis, Harderian gland, intra-orbital lachrymal gland, large intestine wall, liver, pancreas, stomach wall (glandular), trachea, all sub-structures of the adrenal gland, and all tissues in the dermal and fatty systems. Most of the tissues had tissue: plasma AUC0-t ratios greater than 1. Overall, the tissues of the endocrine, fatty, and secretory tissues contained the highest distribution of 14C-emvododstat-derived radioactivity. Little radioactivity was found in central nervous system when compared to most other tissue systems. PK parameters of 14C-emvododstat-derived radioactivity in plasma and tissues are summarised in .
Table 1. Pharmacokinetic parameters of 14C-emvododstat-derived radioactivity in plasma and tissues following a 50 mg/kg 14C-emvododstat oral dose in male Long-Evans rats (n = 1 per timepoint).
AME in rats following a single oral 14C-emvododstat dose
Following a single 40 mg/kg oral dose of 14C-emvododstat in male Sprague Dawley rats, the formulation was well tolerated. The animals appeared to be normal throughout the study and demonstrated no clinical signs from the drug administration.
Following a single 14C-emvododstat oral dose at 40 mg/kg in rats, the mass balance data are summarised in . The representative metabolite profiles are shown in , and the plasma concentration-time curves of TRA, emvododstat, and its metabolites are shown in . Plasma PK parameters and distribution data of emvododstat and its metabolites are listed in and , respectively.
Figure 2. Representative HPLC radiochromatograms of 4 h (A), 24 h (B), 72 h (C) and 168 h (D) plasma, 0–24 h (E) and 24–144 h (F) faeces from intact rats, 0–24 h bile (G), 0–24 h (H), and 24–72 h (I) faeces from bile-duct cannulated rats following a single 40 mg/kg oral dose of 14C-emvododstat.
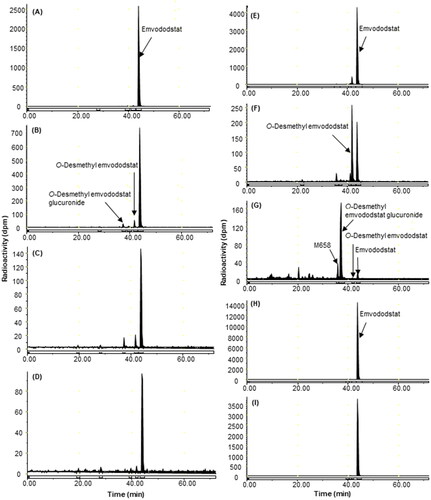
Table 2. Percent of the administered dose recovered following a single oral dose of 14C-emvododstat in male Sprague Dawley rats and male Beagle dogs.
Table 3. Pharmacokinetic parameters for plasma metabolites following a single oral dose of 14C-emvododstat in male Sprague Dawley rats and male Beagle dogs.
Table 4. Percentage of dose excreted as emvododstat and metabolites in pooled bile, urine and faeces following a single oral dose of 14C-emvododstat in male Sprague Dawley rats and male Beagle dogs.
In intact male rats, 58.7% of the 14C-emvododstat-derived radioactivity was excreted by 168 h, and the main route of excretion was through faeces with negligible excretion in urine. For the male BDC rats, 82.4% of the 14C-emvododstat-derived radioactivity was excreted by 72 h, and the main route of excretion was through faeces with minor excretion in bile and urine. The mean Cmax values of 6.65 µg eq/mL in plasma and 4.21 µg eq/mL in blood of 14C-emvododstat-derived radioactivity were both observed at 8 h post-dose. Plasma TRA decreased slowly after Tmax and was detectable (>0.0732 µg eq/mL) by 168 h post-dose. Blood concentrations were lower than plasma concentrations at all time points post-dose and were detectable (>0.141 µg eq/mL) by 168 h post-dose. The blood: plasma concentration ratios from 1 h to 168 h post-dose ranged from 0.57 to 0.79.
Emvododstat, which peaked at 8 h, was the dominant circulating radioactive component in intact rats. O-Desmethyl emvododstat and its glucuronide, both of which peaked at 24 h were the most prominent circulating metabolites. In BDC rat bile, unchanged emvododstat and O-desmethyl emvododstat were minor radioactive components, while O-desmethyl emvododstat glucuronide was the most prominent metabolite. In intact rat faeces, unchanged emvododstat was the most abundant radioactive component and O-desmethyl emvododstat was the most abundant metabolite. In BDC rat faeces, unchanged emvododstat was the dominant radioactive component while O-desmethyl emvododstat was barely detectable (<0.1% dose). Trace amounts (<0.1% dose) of emvododstat and its metabolites were detected in either intact or BDC rat urine.
AME in dogs following a single oral 14C-emvododstat dose
The dose was well tolerated in male beagle dogs following oral administration of 14C-emvododstat at 20 mg/kg. Except for soft stools or light diarrhoea observed during 24 h post-dose, there were no other clinical signs observed till the end of the study for all 3 dogs.
Following a single oral dose of 14C-emvododstat at 20 mg/kg in dogs, the recoveries of dosed radioactivity in urine, faeces, and cage rinse/wash are summarised in . The representative metabolite profiles are shown in , the plasma concentration-time curves of TRA and emvododstat metabolites are shown in . Plasma PK parameters and distribution data of emvododstat and its metabolites are listed in and , respectively.
Figure 3. Representative HPLC radiochromatograms of 4 h (A), 8 h (B), 24 h (C), and 72 h (D) plasma, 0–24 h (E) and 24–48 h (F) faeces from dogs following a single 20 mg/kg oral dose of 14C-emvododstat.
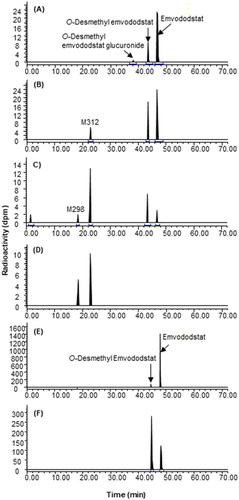
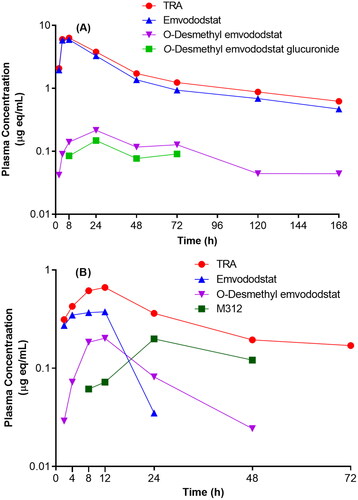
Figure 4. Plasma concentration – time curves of total radioactivity (TRA), emvododstat and its metabolites following oral dose administration in rats (A) and dogs (B).
A total of 92.8% of the dose was recovered by 192 h post-dose and the main route of excretion was through faeces with negligible excretion in urine (1<% dose). The plasma TRA reached Tmax at 8 ∼ 12 h with the mean Cmax of 0.689 µg eq/mL. M312, O-desmethyl emvododstat, and unchanged emvododstat were the major radioactive components in dog plasma. 14C-emvododstat was the dominant component and O-desmethyl emvododstat was the most prominent metabolite in 0–96 h faeces.
Metabolite identification
In addition to unchanged emvododstat, six metabolites were identified or characterised in plasma, urine, or faeces after oral administration of 14C-emvododstat in rats and dogs.
Emvododstat and O-desmethyl emvododstat were identified by direct comparison of HPLC retention times and high-resolution mass spectral data with reference standards. Structures of other metabolites were proposed based on their high-resolution mass spectral data of chlorine isotopic patterns and fragmentation ions relative to emvododstat or O-desmethyl emvododstat. Proposed structures and metabolic pathways for the formation of the detected metabolites are represented in .
Emvododstat
The observed accurate mass for the protonated molecular ion (MH+) of emvododstat was at m/z 467.0924 (calculated 467.0924 with a formula of C25H21O3N2Cl2+). The characteristic product ion at m/z 359.0345 was due to the neutral loss of anisole moiety (C7H6O), and at m/z 121.0647 (C8H9O+) was attributed to 4-methoxyphenylmethylium ion.
O-Desmethyl emvododstat
The observed accurate mass for MH+ of O-desmethyl emvododstat was at m/z 453.0766 (calculated 453.0767 with a formula of C24H19O3N2Cl2+). The characteristic product ion at m/z 359.0346 was due to the neutral loss of phenol moiety (C6H4O), and at m/z 107.0491 (C8H9O+) was attributed to 4-hydroxyphenylmethylium ion.
O-Desmethyl emvododstat glucuronide
The observed accurate mass for MH+ of O-desmethyl emvododstat glucuronide was at m/z 629.1087 (calculated 629.1088 with a formula of C30H27O9N2Cl2+), which is 176.0321 Da (C6H8O6) higher than MH+ of O-desmethyl emvododstat, indicating a glucuronic acid conjugate of O-desmethyl emvododstat. The characteristic product ions at m/z 453.0764, 359.0347, and 107.0491 all agreed well with O-desmethyl emvododstat glucuronide.
M312
The observed accurate mass for MH+ of M312 was at m/z 313.1098 (calculated 313.1102 with a formula of C18H18ON2Cl+), which is 153.9822 Da lower than MH+ of emvododstat, indicating the neutral loss of 4-chlorophenyl formate moiety (C7H3O2Cl) due to amide bond hydrolysis. The characteristic product ions at m/z 205.0530 and 121.0648 also agreed well with the proposed structure.
M298
The observed accurate mass for MH+ of M298 was at m/z 299.0945 (calculated 299.0946 with a formula of C17H16ON2Cl), which is 153.9821 Da (C7H3O2Cl) lower than O-desmethyl emvododstat, or 14.0157 Da (CH2) lower than M312, indicating the neutral loss of 4-chlorophenyl formate moiety due to amide bond hydrolysis from O-desmethyl emvododstat or demethylation from M312. The characteristic product ions at m/z 205.0530 and 107.0492 also agreed well with the proposed structure.
M482
The observed accurate mass for MH+ of M482 was at m/z 483.0875 (calculated 483.0873 with a formula of C25H21O4N2Cl2+), which is 15.9949 Da (1 oxygen) higher than emvododstat, indicating M482 was a mono-oxidation metabolite of emvododstat.
M658
The observed accurate mass for MH+ of M658 was at m/z 659.1194 (calculated 659.1194 with a formula of C31H29O10N2Cl2+), which is 176.0321 Da (C6H8O6) higher than M482, indicating a glucuronic acid conjugate of M482. The characteristic product ions at m/z 483.0869, 312.0784, and 121.0647 also agreed well with the proposed structure.
Discussion
Emvododstat is a highly lipophilic neutral compound with poor aqueous solubility. Following a single oral dose in male Long-Evans rats, distribution of 14C-emvododstat-derived radioactivity was extensive with the endocrine, fatty, and secretory tissues contained the highest radioactivity. The exposure to 14C-emvododstat-derived radioactivity in pigmented skin was higher when compared to the exposure in the non-pigmented skin, but it seemed that the binding was reversible as the concentrations decreased over time (). Consistent with extensive distribution and retention in tissues in male Long-Evans rats, excretion of 14C-emvododstat-derived radioactivity in intact male SD rats was slow, approximately 65% of dose (0.3% in urine, 54.7% in faeces, and 35.4% in carcase) within 7 days post-dose in rats. In contrast, approximately 93% of dose was recovered 8 days post-dose with majority of the radioactivity excreted in 0–24 h faeces in dogs. It should be noted that high radioactivity in cage rinse/wash was largely caused by the diarrhoea and/or soft stools observed in dogs within 24 h post-dose, and by considering this, the actual faecal excretion was close to 80% of dose within 24 h and approximately 92% of dose within 8 days with negligible amount (<1% of dose) excreted in urine following a single oral dose in dogs (). The faster excretion in dogs was most likely due to lower absorption as indicated by the observation that close to 80% of dose was excreted in 0–24 h faeces and >94% of faecal radioactivity was attributed to unchanged emvododstat with little contribution from metabolites. Urinary excretion of 14C-emvododstat-derived radioactivity was low in both rats (0.2% dose within 24 h and 0.3% dose within 7 days post-dose) and dogs (∼0.5% dose within 8 days post oral dose) ().
Absolute oral bioavailability of emvododstat was around 100% following a single oral dose at 3 mg/kg in intact male SD rats (Ma et al. Citation2022), and the plasma exposure (AUC) was close to dose proportional from 4 to 40 mg/kg dose range (internal PTC data). In intact male Long-Evans rats, following a single 50 mg/kg oral dose of 14C-emvododstat, bile in bile duct was one of the tissues with the highest 14C-emvododstat-derived radioactivity (data not shown). However, following a 40 mg/kg oral dose in BDC rats, only 1.96% of dose was recovered in bile with negligible urinary excretion (0.15% of dose). Compared with intact rats following the same dose, excretion rate in BDC rats was faster (82.4% by 72 h in BDC rats vs 58.7% by 168 h in intact rats) and less residual radioactivity remained in the body (18% by 72 h in BDC rats vs 35.4% by 168 h in intact rats). All these observations suggested that low biliary excretion in BDC rats may be largely due to low absorption of 14C-emvododstat-derived radioactivity following oral dose. This was also supported by the metabolite distribution in BDC rats vs intact rats: in BDC rat bile, O-desmethyl emvododstat glucuronide was more abundant (0.63% of dose) than emvododstat (0.05% of dose) and O-desmethyl emvododstat (<0.01% of dose). Based on this, following biliary excretion in intact rats, O-desmethyl emvododstat and O-desmethyl emvododstat glucuronide in intestine are expected to be the main contributors of O-desmethyl emvododstat in faeces. Indeed, compared with BDC rats, much more O-desmethyl emvododstat was detected in faeces from intact rats: 6.54% of dose or approximately 20% of emvododstat in intact rats vs 0.07% of dose or approximately 0.1% of emvododstat in BDC rats (). The combined metabolism and excretion data suggest that emvododstat was well absorbed and the biliary excretion was functioning in intact rats following the 14C-emvododstat oral dose, while in BDC rats the absorption of emvododstat was compromised due to bile canulation. Since emvododstat is highly lipophilic in nature, it is reasonable to assume that the bile salts, the essential components for lipid absorption (Holm et al. Citation2013; Kester Citation2014), are also needed for emvododstat absorption following oral dose administration.
Following a 40 mg/kg single oral dose of 14C-emvododstat in rats, emvododstat was the dominant circulating component while O-desmethyl emvododstat and its glucuronide were the minor metabolites, accounting for 84.5%, 5.48% and 2.22% of plasma TRA, respectively ( and ). In contrast, emvododstat metabolism in dogs was more extensive: emvododstat and 2 metabolites, O-desmethyl emvododstat and M312 were the major circulating components, accounting for 24.6%, 18.5% and 39.1% of plasma TRA, respectively, following a 20 mg/kg single oral dose of 14C-emvododstat in dogs ( and ). Very similar AUC ratios of O-desmethyl emvododstat to emvododstat in each species were observed in this study compared with a previous PK study following 3 mg/kg oral dose in rats and dogs (Ma et al. Citation2022).
The metabolic pathways of emvododstat in rats and dogs are slightly different (). Although O-demethylation followed by glucuronidation are the common pathways in both species, the N-carbamoyl ester link was stable in rats while labile in dogs: hydrolysis on amide side led to the formation of M312 and subsequently M298 in dogs. It also appears that N-carbamoyl amide link cleavage was a slow process since these metabolites were observed at the later timepoints in dogs following oral dose ().
The doses used in the current study were based on the no-observed-adverse-effect level (Gao et al. Citation2013) established in toxicology studies in Sprague Dawley rats and Beagle dogs following repeat daily oral dosing of emvododstat (PTC internal data). The results obtained in this study are essential to compare metabolism and disposition differences across species and to confirm that no unique or disproportionate metabolites in humans after oral dose of emvododstat (U.S. Department of Health and Human Services, Food and Drug Administration Centre for Drug Evaluation and Research Citation2020).
Conclusion
In conclusion, emvododstat is a highly lipophilic compound with extensive tissue distribution in rats following oral dose administration. Metabolism and excretion of emvododstat was species-dependent following a single oral dose in rats and dogs. Excretion of 14C-emvododstat-derived radioactivity was faster in dogs than in rats, while urinary excretion was minimal (<1% of dose) in both rats and dogs.
Emvododstat was the dominant radioactive component in rat plasma and faeces. Emvododstat was also the dominant radioactive component in dog faeces, while emvododstat and two of its metabolites (O-desmethyl emvododstat and M312) were the major circulating components in dog plasma.
Author contributions
Ma J. and Kong R. participated in the research design. Ma J., Ye Q., Babiak J., Northcutt V., and Kong R. conducted experiments, performed data analysis and wrote or contributed to the writing of the manuscript.
Disclosure statement
All authors are current employees except that Babiak J. and Northcutt V. are former employees of PTC Therapeutics, Inc.
Additional information
Funding
References
- Branstrom A, Cao L, Furia B, Trotta C, Santaguida M, Graci JD, Colacino JM, Ray B, Li W, Sheedy J, et al. 2022. Emvododstat, a potent dihydroorotate dehydrogenase inhibitor, is effective in preclinical models of acute myeloid leukemia. Front Oncol. 12:832816.
- Cao L, Weetall M, Bombard J, Qi H, Arasu T, Lennox W, Hedrick J, Sheedy J, Risher N, Brooks PC, et al. 2016. Discovery of novel small molecule inhibitors of VEGF expression in tumour cells using a cell-based high throughput screening platform. PLoS One. 11(12):e0168366.
- Cao L, Weetall M, Trotta C, Cintron K, Ma J, Kim MJ, Furia B, Romfo C, Graci JD, Li W, et al. 2019. Targeting of hematologic malignancies with PTC299, a novel potent inhibitor of dihydroorotate dehydrogenase with favourable pharmaceutical properties. Mol Cancer Ther. 18(1):3–16.
- Gao H, Jacobs A, White RE, Booth BP, Obach RS. 2013. Meeting report: metabolites in safety testing (MIST) symposium – safety assessment of human metabolites: What’s REALLY necessary to ascertain exposure coverage in safety tests? Aaps J. 15(4):970–973.
- Holm R, Müllertz A, Mu H. 2013. Bile salts and their importance for drug absorption. Int J Pharm. 453(1):44–55.
- Ignacio RAB, Lee JY, Rudek MA, Dittmer DP, Ambinder RF, Krown SE. 2016. A phase 1b/pharmacokinetic trial of PTC299, a novel post transcriptional VEGF inhibitor, for AIDS-related Kaposi’s sarcoma: AIDS Malignancy Consortium trial 059. J Acquir Immune Defic Syndr. 72 (1):52–57.
- Kester JE. 2014. Liver. In: Wexler P, editor. Encyclopedia of toxicology. 3rd ed. London (UK): Academic Press; p. 96–106.
- Luban J, Sattler R, Mühlberger E, Graci JD, Cao L, Weetall M, Trotta C, Colacino JM, Bavari S, Strambio-DeCastillia C, et al. 2021. The DHODH inhibitor PTC299 arrests SARS-CoV-2 replication and suppresses induction of inflammatory cytokines. Virus Res. 292:198246.
- Ma J, Kaushik D, Yeh S, Northcutt V, Babiak J, Risher N, Weetall M, Moon YC, Welch EM, Molony L, et al. 2022. In vitro metabolism, pharmacokinetics and drug interaction potentials of emvododstat, a DHODH inhibitor. Xenobiotica. 52(2):152–164.
- Packer RJ, Rood BR, Turner DC, Stewart CF, Fisher M, Smith C, Young-Pouissant T, Goldman S, Lulla R, Banerjee A, et al. 2015. Phase 1 and pharmacokinetic trial of PTC299 in paediatric patients with refractory or recurrent central nervous system tumours: a PBTC study. J Neurooncol. 121(1):217–224.
- U.S. Department of Health and Human Services, Food and Drug Administration, Centre for Drug Evaluation and Research. 2020. Guidance for Industry: safety Testing of Drug Metabolites. March 2020.
- Weetall M, Davis T, Elfring G, Northcutt V, Cao L, Moon YC, Riebling P, Dali M, Hirawat S, Babiak J, et al. 2016. Phase 1 study of safety tolerability and pharmacokinetics of PTC299, an inhibitor of stress-regulated protein translation. Clin Pharmacol Drug Dev. 5(4):296–305.