

Abstract
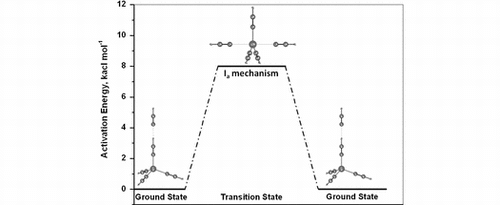
Density functional (B3LYP, B3PW91, X3LYP, BP86, PBEPBE, PW91PW91, and M06) and ab initio (MP2, MP4sdq, CCSD, and CCSD(T)) calculations with extended basis sets (6-311+G**, TZVP, LANL2DZ+p, and SDD+p, the latter including extra polarization and diffuse functions) indicate that HCN exchange on [Cu(HCN)4]+ proceeds via an associative interchange (Ia) mechanism and a D3h transition structure {[Cu(HCN)5]+}‡. The activation barrier, relative to the model complex [Cu(HCN)4]+·HCN, varies modestly, depending on the computational level. Typical values are 8.0 kcal M−1 (B3LYP/6-311+G**), 6.0 kcal M−1 (M06/6-311+G**), and 4.8 kcal M−1 (CCSD(T)/6-311+G**//MP2(full)/6-311+G**). Inclusion of an implicit solvent model (B3LYP(CPCM)/6-311+G**) leads to an activation barrier of 5.8 kcal mol−1. Comparison of the HCN exchange mechanisms on [Li(HCN)4]+ (limiting associative, A) and [Cu(HCN)4]+ (associative interchange, Ia) reveals that π back donation in the equatorial Cu–N bonds in the transition state determines the mechanism.
Graphical Abstract
Computed structural and energetic data indicate an associative interchange (Ia) mechanism for HCN exchange on [Cu(HCN)4]+.
Acknowledgments
The authors gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft. We thank Prof. Tim Clark for hosting this work at the CCC and the Regionales Rechenzentrum Erlangen (RRZE) for a generous allotment of computer time. BMA thanks the Al-Balqa Applied University for their support. MW thanks Prof. Dirk Zahn for continued support.
Notes
In memoriam Prof. Dr Wolf Broda (31.12.1917–4.6.2014).