
Abstract
Despite the proliferation of tungstate and molybdate polyoxometalates (POMs) the inclusion of sulfides into these structures proved to be challenging. The use of appropriate preformed thiomolybdate building blocks has over recent years developed into an extensive field with a wide range of diverse structural features and sizes. Initial explorations focused largely either on a combinatorial approach of POM and thiomolybdate dimeric units or a self-condensation approach which gave ring-shaped structures. Over the past ten years these structural archetypes have been challenged; new design approaches led to the generation of fundamentally new building blocks and multicomponent systems featuring well-defined structural fragments which can self-organize in numerous different ways forming a wide variety of diverse structural features, often of remarkably high nuclearity. The unique properties and high tunability potential of the system has proven valuable, with examples of POTM systems exhibiting highly diverse structural motifs and highly promising properties in catalytic applications. In this mini-review we describe the methodologies that have been used to construct materials with important catalytic and structural properties. The discussion does not aim to provide an exhaustive review of the field but rather focuses on the recent developments that occurred the last decade.
Graphical Abstract
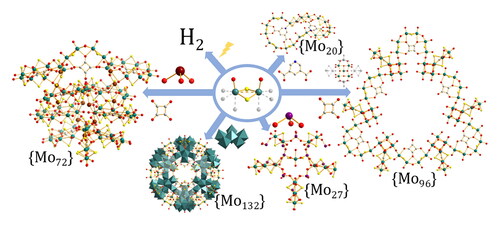
1. Introduction
Metal sulfides play an important role as catalysts both in industrial catalysis [Citation1] and in enzymes [Citation2]. Of particular interest MoS2 presents itself as one of the most promising hydrogen evolution reaction (HER) catalysts for the electrochemical reduction of water to hydrogen as part of a hydrogen economy [Citation3–7], whilst at the same time Mo-S bonds are found in the active sites of bacterial nitrogenases and hydrogenases [Citation8]. It is unsurprising that the formation of complex architectures containing Mo-S bonds would be of interest to chemists either as chemically precise models for MoS2 catalysts, or as artificial mimics of the metal clusters in molybdoenzymes.
Molybdenum can form highly complex molecular species built from alternating MoVI cations and oxo anions known as polyoxometalates (POMs) [Citation9–11]. The replacement of one or more oxo anions in such a structure with sulfide anions would seem a straightforward route to achieving the incorporation of Mo-S bonds into complex molecular architectures to give so called polyoxothiometalates (POTMs). The introduction of sulfide anions, however, is limited by the tendency of sulfide to reduce the do Mo and W metals that are present in these POMs. Success has been achieved using heteroatoms such as in the Keggin structure α-[PW11NbSO39]4– [Citation12] in which sulfide is a terminal ligand of the Nb heteroatom, and the Lindquist structures [W5TaSO18]3– and [W5NbSO18]3– [Citation13] where the sulfide links to Ta and Nb, respectively. The formation of stable bonds directly to Mo and W centers, however, requires higher oxidation states of the metal to avoid reduction by sulfide. This greatly changes the bonding, and hence structure of POTMs built primarily with MoV and MoIV compared to MoVI, WVI and VV. Despite this, structural diversity has been discovered within the family of POTMs, featuring distinct structural motifs compared to POMs.
Rather than direct condensation of individual metal ions found in POMs, POTMs are generally built from preformed thiometalate building blocks. The requirement for a high degree of stability to function as a building block along with multiple electrophilic sites for coordination has thus far limited POTMs to a few building blocks. By far the most common is the dimeric [MoV2O2(µ-S)2]2+ [Citation14, Citation15] (POTM dimer), whose tungstate equivalent [WV2O2(µ-S)2]2+ [Citation16, Citation17] has been less explored due to its instability in water. Lastly, [MoIV3(µ3-S)(µ-S)]4+ [Citation18–20] has also proven to act as a building block in the assembly of POTMs; however, it does not self-condense in the same manner as [MoV2O2(µ-S)2]2+ and therefore has found far less use ().
Figure 1. Structures of the three build blocks used in POTMs. a) [MoV2O2(µ-S)2]2+, b) [WV2O2(µ-S)2]2+ and c) [MoIV3(µ3-S)(µ-S)]4+. Mo, teal; W, blue; O, red; S yellow; open coordination faces highlighted with translucent grey atoms.
![Figure 1. Structures of the three build blocks used in POTMs. a) [MoV2O2(µ-S)2]2+, b) [WV2O2(µ-S)2]2+ and c) [MoIV3(µ3-S)(µ-S)]4+. Mo, teal; W, blue; O, red; S yellow; open coordination faces highlighted with translucent grey atoms.](/cms/asset/33514f31-6009-4154-86e0-c7e9a6492ce7/gcoo_a_2086049_f0001_c.jpg)
[MoV2O2(µ-S)2]2+ consists of two MoV cations, bridged by two sulfide anions and possessing two terminal oxo anions, one attached to each MoV. Both oxo anions lie on the same side of the molecule, giving overall C2v symmetry, and the oxo anions are tilted approximately 18° away from each other, giving the overall cation a slight bend [Citation21]. This gives a tendency to form closed atomically precise structures, rather than extended oligomers. Each MoV possesses three uncoordinated faces for an octahedral geometry, which are occupied by labile solvent ligands in solution, and present easy points of attachment to constructing POTMs. The equatorial positions (relative to the oxo ligand) are more available due to lower steric hindrance, the axial position is both more sterically crowded and less electronically favorable due to the trans oxo ligand. The MoV cations are separated by 2.8 Å, giving a formal single metal-metal bond occupied by the single electrons present on each molybdenum; this lends stability to the building block, despite the V oxidation state which is unusual for molybdenum otherwise. [WV2O2(µ-S)2]2+ possesses the same structure, with tungsten in place of molybdenum.
[MoIV3(µ3-S)(µ-S)]4+ conversely contains molybdenum in a IV oxidation state. It has overall C3V symmetry, with three MoIV in an equilateral triangle, bridged on one side with a µ3 sulfide anion, and on the other side with three µ sulfide anions [Citation22]. The MoIV cations are separated by 2.7 Å, and possess three open coordination faces, each lying approximately opposite one sulfide ligand.
These building blocks can then be linked together to form larger POTMs featuring multiple copies of the same building block. Their construction has been approached generally in one of two ways, leading to two distinct families of POTMs.
The first approach is to link lacunary POMs with POTM building blocks. Lacunary POMs are those which are “missing” one or more metal atoms compared to a “complete” POM structural motif. These vacant sites are highly nucleophilic, and perfectly sized to fit the electrophilic Mo cations in the building blocks, substituting whichever solvent ligands are present. This leaves one ([MoV2O2(µ-S)2]2+, [WV2O2(µ-S)2]2+) or two ([MoIV3(µ3-S)(µ-S)]4+) uncoordinated molybdenum cations on the other side of the POTM building block. When possible these may coordinate in an adjacent lacunary site giving a monomeric structure [Citation23] but more commonly will coordinate to a second lacunary POM in an effort to achieve enhanced structural stability. This creates structures consisting of multiple POM fragments, each linked by bridging POTM building blocks varying in nuclearity from 2-6. The synthetic procedures simply involve mixing solutions of preformed lacunary POM and POTM building blocks together, however, they are highly sensitive to specific reaction conditions, with different synthetic conditions leading to the formation of different structures, while the choice of cation can introduce further structural stability via a template effect [Citation24].
The second method is to self-condense [MoV2O2(µ-S)2]2+ units (or [WV2O2(µ-S)2]2+) into oligomeric rings (). Although [MoIV3(µ3-S)(µ-S)]4+ is able to dimerize [Citation25] it does not self-condense in the same way as the other building blocks, and is thus excluded from this family of POTMs. Oligomerization is initiated by raising the pH of an aqueous solution of [MoV2O2(µ-S)2(H2O)6]2+ leading to deprotonation of one of the aqua ligands to hydroxide, which is then able to bridge to a second [MoV2O2(µ-S)2(H2O)6]2+ unit, substituting another aqua ligand. This occurs multiple times, eliminating all of the aqua ligands in equatorial positions, and replacing them with bridging µ-hydroxide ligands. The aqua ligands in axial positions likewise also start bridging, each coordinating to the axial face of MoV on two different building blocks but are not deprotonated. This leads to an oligomeric “ribbon” structure of general formula [MoV2O2(µ-S)2(µ-OH)2(µ-H2O)]n which notably holds no overall charge. Rather than form extended polymeric chains, however, these oligomers form closed loops, assisted by the inherent curve in the structure of each [MoV2O2(µ-S)2]2+ sub-unit.
Figure 2. Schematic representation of the self-condensation process of [MoV2O2(µ-S)2(H2O)6]2+ as a result of increased pH.
![Figure 2. Schematic representation of the self-condensation process of [MoV2O2(µ-S)2(H2O)6]2+ as a result of increased pH.](/cms/asset/16517a57-33f7-4e0d-9bdf-536d1bacb5b7/gcoo_a_2086049_f0002_c.jpg)
In the absence of any structure directing factors [MoV2O2(µ-S)2(µ-OH)2(µ-H2O)]6 is formed, however, the structure and nuclearity can be directly controlled using a template-driven assembly approach. In the presence of iodides which hydrogen bond to the internal water the contracted n = 5 structure [(MoV2O2(µ-S)2(µ-OH)2(µ-H2O))5I2]2– is obtained to better fit the size of the iodide anion [Citation26]. Due to the presence of iodide as part of the synthesis of [MoV2O2(µ-S)2(H2O)6]2+ this is a commonly obtained structure, often hydrolyzed in acid as a starting material for other POTMs. The internal aqua ligands may also be replaced with multidentate oxygen-bearing anions which act as structural templates. Rather than forming perfect circles the POTM ribbon wraps around one or two such templates, allowing the oxygen atoms on the template to coordinate to the axial positions of the molybdenum, essentially forming a host-guest system. A wide variety of templates have been used, including both rigid and flexible organic poly-carboxylates [Citation27–34], inorganic anions [Citation35–39] and coordination compounds [Citation40, Citation41]. As a host-guest system the host POTM is highly adaptable and will change nuclearity in order to accommodate whichever guests are present. In order to describe structures with more clarity a shorthand is often adopted of the form {MonTempm} for a ring with nuclearity of n MoV and containing m templates. Where it adds clarity additional components may also be shown to emphasize the total number of sulfur, or internal aqua ligands. Overall charge is ignored.
The purpose of this mini review is to highlight the recent developments in the field observed in the last decade following the excellent reviews reported by Cadot et al. in 2011 [Citation42] and 2012 [Citation43], rather than an exhaustive review of the POTM field. In particular a focus on new approaches giving rise to structures which are hybrids of both POTM families, and those with bonding distinct from these two families, of which very few examples exist before 2012 [Citation44–46].
2. Further development of rings
“Traditional” POTM rings, those following exactly the bonding modes and structure types described earlier, have continued to be explored. Many questions regarding their properties still remain, including how to predict stability of the template-ring host-guest arrangement, how the electrocatalytic properties may be optimized, and how to predict and control the outcome in cases where multiple possible templating geometries are possible. Recent publications have helped to shed some light on these questions.
Floquet and Cadot et al. examined how templates may be modified to improve thermodynamic stability. They noted that the hexadecameric structure {Mo16(IsoP)2(H2O)2}4– (IsoP = isophthalate) which is doubly templated by isophthalate ligands features the two hydrogens in the 3 positions of the templates in a close approximation to the geometry of an azo bond. A single template consisting of two isophthalates linked by an azo bond (giving 3,5-dicarboxyl-(3′,5′-dicarboxyazophenyl)benzene (abtc4–)) would therefore be nearly isomorphic to the isophthalate complex. With this ligand they were able to synthesize the hexadecameric structure {Mo16(abtc)(H2O)2}4–, but surprisingly were also able to isolate the doubly templated {Mo16(abtc)2(H2O)2}8– with a very high charge/Mo ratio of 0.5 e per Mo [Citation47].
The stability in solution could be assessed using NMR. A considerable downshift of resonances coming from the ligand is observed due to the presence of the POTM ring, which makes the trace distinct from that of the solvated ligand. The relative ratios could then be compared to give a value for the equilibrium between solvated and complexed ligands. The singly templated form was completely stable in both D2O and d6-DMSO exhibiting no quantitative amount of solvated ligand. The doubly templated form would instantly convert to the singly templated form, releasing exactly one equivalent of the template in both D2O and d6-DMSO. When two equivalents of IsoP2– were added and allowed to equilibrate the abtc4– remained complexed, whilst IsoP2– remained completely solvated, indicating significant increase in stability in the linked form of the template. This is attributed to the chelate effect, along with the greater rigidity of the abtc4– template. These results confirm observations that high number of attachment points and highly rigid templates lead to greater stabilities.
The squarate template (C4O4) has been demonstrated by Cronin, Miras et al. as a method to access a very wide range of POTM structures (). The full range of structures available in conjunction with other linking units will be described later. Even on its own, however, it has two distinct templating geometries leading to multiple possible structures. In both cases it templates an 180° “turn” of the [MoV2O2(µ-S)2(µ-OH)2]n chain, however, this may be accomplished with two or three [MoV2O2(µ-S)2(µ-OH)2] units. These “second order building blocks” are a central feature of squarate-POTM chemistry. No consistent nomenclature exists to describe these building blocks, so for the sake of clarity they will be termed “square-type” and “diamond-type” based on the orientation of the squarate unit when the units open coordination faces are aligned horizontally. In the “square-type” configuration three units surround each squarate, at right angles to each other forming three sides of a square, mimicking the arrangement of the C4 square of the template. The squarate points an oxygen towards each corner of the square. Each unit coordinates to two of the oxygen atoms on the squarate, one in the axial position of each MoV. In the “diamond-type” configuration two units are used and the template is rotated 45° relative to the square configuration. Both units coordinate to two oxygens each, as in the square configuration, however, in the diamond-type structure this leaves oxygen atoms from the squarate pointing towards the open coordination faces. Commonly these will coordinate to two further units which are a 45° rotation from the adjacent units, and coordinate with only one MoV to one oxygen, leaving the other MoV available to coordinate to an aqua ligand. These outer units are less strongly coordinated, and have a greater degree of flexibility, adapting to the structural demands of other elements of the structure and being absent in many structures. Interestingly the first reported structure templated by squarate featured both second order building blocks, giving an overall {Mo14(C4O4)2} structure [Citation48]. In the presence of increased concentration of K+, however, a new product became favored. This {Mo16(C4O4)2K} ring featured two squarate templates, both now in square-type configurations. Two additional [MoV2O2(µ-S)2(µ-OH)2] complexes fit between these two, and featured four aqua ligands which coordinated also to a pair of central K+ ions which induced the change in configuration [Citation49]. Remarkably the removal of previously formed {Mo14} was sufficient to raise the concentration of K+ such that {Mo16} became the favored product. A similar treatment was attempted using acetate as a template, whose small size and single point of attachment should make it ideal for adopting multiple possible configurations. Despite this only a single structure, featuring a {Mo12} ring exhibiting an ellipsoid shape and containing two acetate ligands, could be isolated.
Figure 3. Crystal structures of the POTM rings described in this section. A) {Mo16(IsoP)2}, {Mo16(abtc)}, {Mo16(abtc)2}; B) {Mo14(C4O4)2}, {Mo16(C4O4)2K2}, {Mo12(AcO)2}; C) {Mo10}n{thiophenediphosphonate}n; D) {Mo16(DFMT)}, {Mo12(DMT)}. (IsoP = isopthalate, abtc = 3,5-dicarboxyl-(3’,5’-dicarboxyazophenyl), DFMT = 2,5-bis(trifluoro)methylterephthalate, DMT = 2,5-dimethylterephthalate). Formulas are given in shorthand for clarity. Mo, teal; N, blue; O, Red; P, purple; C, white; S, yellow; K, grey; F, green.
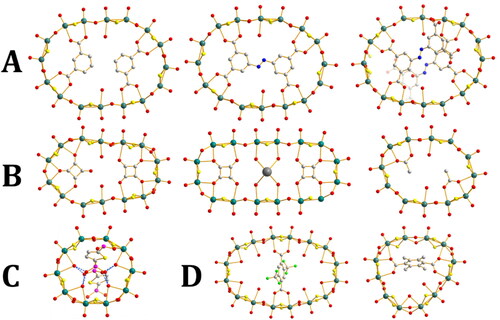
Konchenko et al. were able to modify the {Mo10} ring with a structure directing ligand than lies outside of the ring [Citation50]. By adding thiophenediphosphonate to [(MoV2O2(µ-S)2(µ-OH)2(µ-H2O))5I2]2– the external iodide anions were replaced with the phosphonate groups, which were hydrogen bonded to the internal water molecules of the ring. This prompted a modification of the ring shape away from a perfect circle, and also led to the loss of one internal water molecule. At the same time each diphosphonate was able to link between two rings, creating a 1-D chain of alternating rings and diphosphonates.
Cadot et al. synthesized a range of rings using benzene-based polycarboxylic acids in order to investigate how structural changes could influence the electrocatalytic activity observed from the rings [Citation51]. The terephthalate derivatives, tetramethyterephthalate, 2,5-dimethylterephthalate and 2,5-bis(trifluoro)methylterephthalate, were all used as templates and compared to benzene tricarboxylate as a template. 2,5-Dimethylterephthalate gave an {Mo12(DMT)} (DMT = 2,5-dimethylterephthalate) ring with the same structure as the previously known tetramethylterephthalate in which the benzene ring is twisted out of the plane of the thiomolybdate ring due to steric restrictions. 2,5-Bis(trifluoro)methylterephthalate, however, forms two distinct structures. The first is assumed to be an analogous {Mo12(DFMT)} (DFMT = 2,5-bis(trifluoro)methylterephthalate) structure based on NMR studies but could not be isolated as suitable single crystals. The second is {Mo16(DFMT)} based on a single template, with the ring forming an ellipse shape, with the unique structure being recorded from single crystals. These structures, in addition to the previously known structures based on similar templates, were compared in terms of electrocatalytic ability, the results of which will be discussed in a later section detailing the developments made in electrocatalytic properties.
3. New structural features in POTMs
A diversity of new structures whose bonding does not fit neatly into either the POM-linking or self-condensation category has been discovered. This includes self-condensation/POM fragment hybrids and [MoV2O2(µ-S)2]2+ dimer units linked not by hydroxide but by intentionally added ligands with the ability to act as templates. This presents new horizons in the development of POTMs with novel structural motifs. With multiple bonding modes available, and substructures that may fit together in multiple ways, it however becomes far harder to predict the synthetic outcomes of any particular conditions, or indeed which conditions may lead to novel outcomes. It is therefore necessary to explore a vast combinatorial space in order to discover such structures.
Cronin, Miras et al. found a great deal of success by adopting an autonomous screening system based on a flow system which was controlled algorithmically [Citation52]. They were also able to use conditions known to give desirable structural fragments based on known structures, which decreased the chemical space being searched. The combination of high throughput synthesis along with an understanding of the structural fragments promoted by different conditions and well-designed algorithms is important for the discovery of multicomponent structures such as many of the ones described in this section.
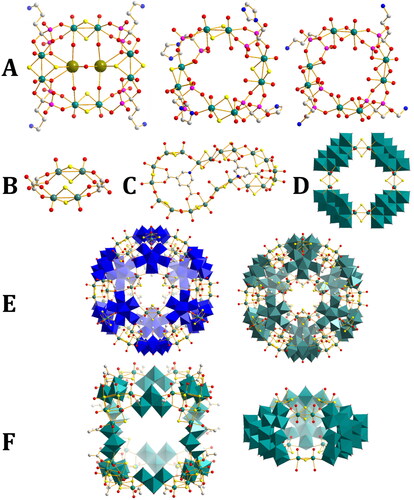
The use of bridging ligands other than hydroxides can have a marked impact on structure. By using the organic bisphosphonates, zoledronic acid (Zol) and alendronic (Ale) acid, Dolbecq et al. were able to construct POTM rings in non-flat geometries [Citation53]. In these structures the phosphonate groups of the bisphosphonates bridge between different [MoV2O2(µ-S)2]2+ groups, each phosphonate connected to each MoV center with a different oxygen atom, and the adjacent deprotonated hydroxy group of the bisphosphonate coordinates to one of the MoV ions, giving an overall pentadentate ligand. Both structures from Zol and Ale consist of four [MoV2O2(µ-S)2]2+ groups linked by bisphosphonates. With Zol the ligands point alternatively up and down, giving an overall S4 symmetry, whereas with Ale all ligands point down, with half the [MoV2O2(µ-S)2]2+ units pointing up and half pointing down, with an overall C2v symmetry while a Rb+ cation is trapped inside the ring, disordered over two positions. Interestingly when Ale is combined with a solution of reduced molybdate a structure is found with the same core as {Mo8S8Zol4}, excepting the replacement of S with O giving a [MoV2O2(µ-O)2]2+ unit. Despite containing Ale, this is different to {Mo8S8Ale4} [Citation54].
Zhang and Xin et al. were able to form {Mo4(Tart)2}-based (Tart = tartarate) clusters using racemic tartaric acid as a bridging ligand instead of hydroxide [Citation55]. Each end of the tartarate ligands binds in an η2 manner through one carboxylate oxygen and one hydroxy oxygen. Both L and D forms are present in each cluster, meaning the overall cluster is non-chiral. Because the tartarate ligand coordinates both dimeric units on a single side only two dimeric units are needed to form a complete ring, making this the lowest possible nuclearity POTM ring, whilst the purely hydroxide bridged forms show at minimum four dimer units (eight total molybdenum ions).
Recently Zhang and Xin et al. used the nitrogen-bearing ligand 3,5-pyridinedicarboxylate (PyD) as a template [Citation56]. Instead of giving a flat {Mo16PyD2} ring as would be expected for isophthalate (isostructural save for the exchange of a benzene ring with a pyridine ring) they found a {Mo20PyD2} ring whose structure consisted of a large loop with a pinch in the middle wherein two [MoV2O2(µ-S)2]2+ units have rotated such that their terminal oxo groups point inwards. This gives two partial loops, each wrapping around one PyD2– template, which are then twisted relative to each other. This distinctive geometry is prompted by the coordination of nitrogen on the PyD template to the Mo on one of the rotated [MoV2O2(µ-S)2]2+ groups. The nitrogen is much closer to the center of the template than the carboxylate group and disrupts the formation of an entirely convex ring as seen in {Mo16IsoP2}. The propeller-like twist makes the overall structure chiral, however, it crystallizes in a unit cell that contains both enantiomers.
The combination of lacunary POMs with [MoV2O2(µ-S)2]2+ has previously been limited to those POM fragments which can be isolated with lacunary sites; however, it is known that many virtual POM-based building blocks exist in solution. Although they cannot be isolated directly, the repeated motifs can be observed in larger POMs. These often have several nucleophilic coordination points suitable for [MoV2O2(µ-S)2]2+ and therefore can be used to synthesize a wide variety of new structures consisting of virtual building blocks linked by [MoV2O2(µ-S)2]2+.
In 2010 Cadot et al. were able to isolate an {Mo8O28} fragment from an aqueous solution of molybdate with [MoV2O2(µ-S)2]2+ to give a tetrameric structure [(Mo8O28)4(MoV2O2(µ-S)2)4]24– demonstrating the effectiveness of this technique () [Citation57].
Figure 4. Crystal structures of the POTMs described in this section. A) {Mo8S8(Ale)4}, {Mo8S8(Zol)4}, {Mo8O8(Ale)4}; B) {Mo4(Tart)2}; C) {Mo20(PyD)2}; D) {MoV8S8(MoVI8)4}; E) {WVI72MoV60S60}, {MoVI72MoV60S60}; F) {(MoV6S6)8(MoVI3)12}, {(MoVI17)3MoV12S6} (Ale = alendronic acid, Zol = zoledronic acid, Tart = tartarate (racemic), PyD = 3,5-pyridinedicarboxylic acid). Formulas are given in shorthand for clarity. Mo, teal (MoV, spheres, MoVI polyhedra); W, dark blue polyhedra, N, blue spheres, O, red; C, white; S, yellow; Rb, green; hydrogen omitted for clarity.
One of the well-known virtual building blocks is the pentagonal {M(M5)O21} (M = Mo or W) units. This motif has been observed on other occasions, such as in the high nuclearity {Mo132} “Keplerate” structure consisting of pentagonal units linked by [MoV2O2(µ-O)2]2+ dimers to form a truncated icosahedron which is internally lined with acetate ligands [Citation58]. It is unsurprising then that the linking dimer could be swapped for [MoV2O2(µ-S)2]2+ to give a Keplerate-type POTM ().
In 2011 Müller et al. constructed such a structure using tungstate pentagonal units to give {W72Mo60S60} [Citation59]. Following this in 2012 Cadot et al. isolated the pure molybdenum equivalent {Mo132S60} [Citation60]. Compared to the standard Keplerate structure the introduction of sulfides to the edges of the large hexagonal pores would be expected to change their character, and thus the ability of the Keplerate to act as a host for guests. The larger sulfide anions would restrict pore size, while also softening the pore compared to oxo anions. From the same reaction conditions Cadot et al. were also able to isolate a nanosized cubic cluster {Mo84S48}. This consisted of eight [(MoV2O2(µ-S)2)3(µ3-OH)]5+ triangular units linked by (MoVI3O11)4– POM fragments in a cubic arrangement. The unique triangular units represent a very different method of condensation for the [MoV2O2(µ-S)2]2+ unit. The units possess C3 symmetry, and half the bridging sulfides also coordinate to a neighboring MoV, adopting a pseudo-µ3 coordination mode. A singular hydroxide bridge links all three units. Acetate units lie on the outside of this structure, rather than the inside, and for this reason the structure was termed an “inverse Keplerate.” In this way the authors could capture two distinct building blocks from solution using [MoV2O2(µ-S)2]2+, the {Mo6O21}6– (Keplerate) and the {Mo3O11}4– (inverse Keplerate) which exist in equilibrium. This equilibrium is sensitive to pH, with {Mo3O11}4– sufficiently favored by high pH that Keplerate formation is completely avoided with a pH increase of only 0.3 units from the original conditions used.
Further studies exchanged the acetate ligand which supported the Keplerate structure for sulfate ligands, which are also known to support such a structure [Citation61]. In addition to the expected Keplerate structure, a second smaller cage-type structure in the family of {Mo57M6} structures [Citation62] was obtained from diluted solutions under the same conditions. These featured three larger POM fragments {Mo17O52(OH)4(H2O)6} formed primarily from the linking of two pentagonal units to give a horseshoe shape which may be seen as half of the {Mo36} structure [Citation63].
Each end of these “horseshoes” connect to two [MoV2O2(µ-S)2]2+ units, one at each end, giving a pair of triangular pores with the same structure as the pores found in the full sized Keplerates. These pores lie opposite each other, with three differently shaped pores being formed by the gaps between the POM fragments. This structure could act as host to a NMe+ cation in the central cavity, and further featured three strongly associated NMe+ cations in the peripheral spaces between the POM fragments.
3.1. Molybdate/squarate systems
The squarate system continues to show a diversity of possible structures. This may be partly attributed to the fact that whilst it strongly templates a 180° turn in two different coordination modes (square-type and diamond-type, as described earlier), these substructures do not directly dimerize with themselves. Instead in the absence of other elements, rather than form dimers with themselves, an odd {Mo14} ring with both different coordination modes is formed, even though the widths of the two substructures do not well match. At the same time a theoretical {Mo8(C4O4)} is sterically infeasible due to squarate being slightly too large to fit easily; this is in comparison to the slightly smaller oxalate template that forms highly stable {Mo8(Ox)} clusters [Citation27]. The squarate template on its own therefore lacks any thermodynamic “traps” which feature closed systems with no open coordination faces. In this way the addition of squarate creates larger substructures in solution that may be treated as “second order building blocks,” that is building blocks made from other building blocks. Each possess two open coordination faces with a horseshoe-like arrangement. These can then give rise to a wide variety of structures, based on what other components are introduced into solution.
The addition of molybdate (MoVI) was found to lead to several interesting new structures [Citation48]. A dimeric [MoVI2O8]4– unit with two µ-O2– forms at pH 5 and acts as a doubly bidentate linker between [MoV2O2(µ-S)2]2+ units. In the absence of molybdate a {Mo14(C4O4)2} ring consisting of both square-type and diamond-type second order building blocks (described earlier) is formed. When molybdate is added a {Mo12(C4O4)2(MoVI2O8)2} ring is formed made from two square-type building blocks. Notably the attachment points on (MoVI2O8)4– are not directly opposite each other but are offset somewhat. This causes the overall ring to exist on two parallel planes, with the (MoVI2O8)4– moiety acting as a step between the two ().
Figure 5. Building blocks used in squarate/molybdate system. A) [MoV2O2(µ-S)2]2+, B) Squarate ([C4O4]2–), C) Square-type building block, D) Diamond-type building block, E) [MoVI2O8]4–, F) Lindqvist fragment [MoVI5O18]6– top-down and side-on views, G) Pentagonal fragment [MoVI6O21]5– top-down and side-on views. Mo, teal; O, red; S, yellow; C, white; coordination positions shown as translucent grey.
![Figure 5. Building blocks used in squarate/molybdate system. A) [MoV2O2(µ-S)2]2+, B) Squarate ([C4O4]2–), C) Square-type building block, D) Diamond-type building block, E) [MoVI2O8]4–, F) Lindqvist fragment [MoVI5O18]6– top-down and side-on views, G) Pentagonal fragment [MoVI6O21]5– top-down and side-on views. Mo, teal; O, red; S, yellow; C, white; coordination positions shown as translucent grey.](/cms/asset/640d9643-6dda-4102-968e-a7a07fa26c28/gcoo_a_2086049_f0005_c.jpg)
This is however not the only possible way for the (MoVI2O8)4- unit to add to the ring. The increased bridging distance of (MoVI2O8)4– compared to hydroxide bridges makes it possible to complete the theoretical {Mo8(C4O4)} ring (). Because the two coordination faces of (MoVI2O8)4– are offset these change the theoretical ring shape into a circular helix with a very small pitch which, as a helix, introduces structural chirality to the cluster. This is due to the inherent chirality of the staggered coordination modes of (MoVI2O8)4–, however, the presence of two such elements, with opposite handedness, in the larger {Mo12(C4O4)2(MoVI2O8)2} species causes that structure to be non-chiral. The chirality only occurs once the uneven coordination happens, locking in the coordination mode. The building blocks that make up the structure themselves are non-chiral. A non-chiral form could also be isolated, featuring an altered structure of (MoVI2O8)4– with a single µ-O2– and coordination faces directly opposite each other.
Figure 6. Squarate/[MoVI2O8]4– system showing how the building blocks can assemble into A) {MoV12(C4O4)2(MoVI2O8)2}, B) {MoV8(C4O4)(MoVI2O8)} or C) {MoV8(C4O4)(MoVIO4)2}. Mo, teal; O, red; S, yellow; C, white; coordination positions shown as translucent grey.
![Figure 6. Squarate/[MoVI2O8]4– system showing how the building blocks can assemble into A) {MoV12(C4O4)2(MoVI2O8)2}, B) {MoV8(C4O4)(MoVI2O8)} or C) {MoV8(C4O4)(MoVIO4)2}. Mo, teal; O, red; S, yellow; C, white; coordination positions shown as translucent grey.](/cms/asset/356e2c78-3661-48df-a79a-e2392965fe4a/gcoo_a_2086049_f0006_c.jpg)
Larger molybdate fragments in the form of virtual building blocks that exist in solution can be combined with these second order building blocks formed from squarate (). Beyond (MoVI2O8)4– which can coordinate on two faces, and acts as an alternative bridge to hydroxide ligands when larger fragments are used, these present more than two attachment points, allowing the formation of more geometrically complex extended structures. The [MoVI5O18]6– lancunary Lindqvist fragment can be formed and incorporated at lower pH values than the dimer. It presents four positions around the edge to which POTM fragments may attach.
Figure 7. Squarate/[MoVI5O18]6– system showing how the building blocks can assemble into A) {MoV30(C4O4)5(MoVI5O18)3}, B) {MoV32(C4O4)5(MoVI5O18)3}, C) {MoV40(C4O4)7(MoVI5O18)3}, D) {MoV66(C4O4)12(MoVI5O18)6} or E) {MoV48(C4O4)8(MoVI5O18)4}. Mo, teal; O, red; S, yellow; C, white; Coordination positions shown as translucent grey. Red arrows indicate structural relationship and do not necessarily indicate reaction pathway.
![Figure 7. Squarate/[MoVI5O18]6– system showing how the building blocks can assemble into A) {MoV30(C4O4)5(MoVI5O18)3}, B) {MoV32(C4O4)5(MoVI5O18)3}, C) {MoV40(C4O4)7(MoVI5O18)3}, D) {MoV66(C4O4)12(MoVI5O18)6} or E) {MoV48(C4O4)8(MoVI5O18)4}. Mo, teal; O, red; S, yellow; C, white; Coordination positions shown as translucent grey. Red arrows indicate structural relationship and do not necessarily indicate reaction pathway.](/cms/asset/807d2d6d-948f-4594-9e25-977e32e29605/gcoo_a_2086049_f0007_c.jpg)
In general, one pair of opposite attachment sites will point parallel to each other, and directly up from [MoVI5O18]6–. Due to steric considerations the other two attachment points will point away from the central unit, at 90° from each other. Each pair lies on a plane perpendicular to the other. The first two attachment points are well sized to coordinate to both ends of a square-type second order building block forming a pendant loop. The latter two attachment points can alternate with second order squarate building blocks, rotating first 90° one way, then 180° the other for an overall 90° turn. When placed in sequence these form a closed loop of alternating building blocks (the angles described are both approximate and flexible, allowing a wider range of structures to form than just tetramers). Therefore a four component “building block library” is set up, containing [MoV2O2(µ-S)2]2+, [(MoV2O2(µ-S)2)3(OH)4(C4O4)] (square-type), [(MoV2O2(µ-S)2)2(OH)2(C4O4)] (diamond-type) and [MoVI5O18]6– (of note compared to the ring structures the diamond fragment is missing its two outer [MoV2O2(µ-S)2]2+ units).
These elements can be combined in well understood ways, and thereby lead to a whole family of POTMs with nuclearity far greater than those previously reported [Citation64, Citation65]. These structures are selected by varying the relative concentrations of elements along with pH, likewise the only distinction between the conditions giving the (MoVI2O8)4– dimer featured earlier is the use of lower pH.
A total of five different structures were isolated from this system (). The smallest consists of three Lindqvist fragments [MoVI5O18]6– which connect in a chain by two square-type second order building blocks, and each contains a pendant square-type building block. Unlike the other structures the overall cluster does not form a closed loop. The next smallest cluster is achieved when a single [MoV2O2(µ-S)2]2+ serves to close the loop; alternatively a pair of diamond-type building blocks linked together by one [MoV2O2(µ-S)2]2+ unit can also close this loop to give a new structure. The two final structures were isolated from the same conditions, but under the directing effect of a different counter cation. In the presence of K+ a D3h structure {MoV66(MoVI5)6(C4O4)12} is achieved consisting of six [MoVI5O18]6– fragments linked alternatingly by square-type and diamond-type building blocks, and each holding a pendant square building block. In the presence of Cs+ instead a tetrameric structure {MoV48(MoVI5)4(C4O4)8} of D4h symmetry with four [MoVI5O18]6– fragments, each joined by square second order building blocks and each holding a pendant square building block is found instead.
Figure 8. Squarate/[MoVI(MoVI5)O21]5– system and related systems with croconate. Colored circles represent the components present in the conditions that give the indicated product. A) {MoV18(C4O4)9}, B) {MoV10(C5O5)}, C) {MoV2(C5O5)} co-crystallized with {MoVI12(C4O4)4}, D) {MoV36(C4O4)4(MoVI(MoVI5))2}, E) {MoV48(C4O4)6(MoVI(MoVI5))2}, F) {MoV48(C4O4)6(MoVI(MoVI5))2} co-crystallized with {MoVI12(C4O4)4}. Mo, teal; O, red; S, yellow; C, white; coordination positions shown as translucent grey. Red arrows indicate structural relationship and do not necessarily indicate reaction pathway.
![Figure 8. Squarate/[MoVI(MoVI5)O21]5– system and related systems with croconate. Colored circles represent the components present in the conditions that give the indicated product. A) {MoV18(C4O4)9}, B) {MoV10(C5O5)}, C) {MoV2(C5O5)} co-crystallized with {MoVI12(C4O4)4}, D) {MoV36(C4O4)4(MoVI(MoVI5))2}, E) {MoV48(C4O4)6(MoVI(MoVI5))2}, F) {MoV48(C4O4)6(MoVI(MoVI5))2} co-crystallized with {MoVI12(C4O4)4}. Mo, teal; O, red; S, yellow; C, white; coordination positions shown as translucent grey. Red arrows indicate structural relationship and do not necessarily indicate reaction pathway.](/cms/asset/aae2c500-2a1d-4bdb-aa09-9a89218cb9c3/gcoo_a_2086049_f0008_c.jpg)
The Lindqvist fragments are not the only molybdate cluster that can be extracted from solution to pair with squarate-based building blocks. Under similar conditions extended structures can be formed that coordinate to the pentagonal unit {Mo(Mo5)} [Citation52]. The pentagonal unit can coordinate to POTM chains at any position around its edge, and also directly on the face of the pentagon. Up to two pendant square-type second order building blocks can coordinate to each face to form loops. Owing to steric restrictions the formation of more than two POTM chains coordinating to the edge of the pentagon, or to both faces is not observed.
A series of structures based on the parent {Mo48} could be isolated. A closed loop consisting of two pentagonal units connected on either side by a square-type second order building block forms the structure’s core, whilst each pentagon bears two pendant loops. This gives the overall structure a butterfly-like appearance consisting of a body with the pendant loops forming “wings.” This can be modified to {Mo34} by loss of one pendant loop on either side. {Mo48} may co-crystallize with a cluster formed from MoVI and bridging squarates formulated [MoVI12(C4O4)4O36]8– which bridges to {Mo48} via mutual coordination to K+. The differences in conditions that lead to pentagonal-based structures as opposed to Lindqvist-based structures are minor. Both decreased and increased pH can be used, and only a minor increase in [MoV2O2(µ-S)2]2+ concentration is consistent. In the absence of molybdate a smaller {Mo18} cluster consisting of three square-type second order building blocks linked by bridging squarate ligands could also be isolated.
The use of croconic acid (C5O5H2) leads to {Mo10(C5O5)} inevitably being templated, or alternatively a single [MoV2O2(µ-S)2]2+ dimer coordinated on either side to the template ([MoV2O2(µ-S)2(C5O5)2]2–) which also co-crystallized with [MoVI12(C4O4)4O36]8– in the presence of molybdate and squarate. Likewise, the use of tungstate, despite forming similar pentagonal units, would inevitably form {Mo8(WO4)} rings instead. This is an example of competitive templating, where a highly stable closed structure is formed, preventing the aggregation of building blocks into larger structures. It is precisely because such stable, mono-templated structures do not exist for squarate that it shows this ability to form large clusters.
3.2. EO32- and EO32-/squarate systems
Discovering other such templates that form distinct second order building blocks, and avoid exclusively forming closed rings, would provide a similar degree of structural diversity allowing for extended clusters to form from several second order building blocks. EO32– (E = S, Se, Te) anions were found to fit these requirements. They possess multiple oxygens which may coordinate to axial or equatorial positions of [MoV2O2(µ-S)2]2+ along with a lone pair of the central chalcogenide which may further coordinate. However, unlike the tetrahedral inorganic anions they do not generally template closed rings. With a wide variety of coordination modes, the ability to coordinate to more than two units simultaneously and a lack of a single highly stable structure to act as a thermodynamic “sink” these ligands are in a perfect position to generate a family of structurally complex clusters.
Sulfite has been used numerous times in combination with POMs to generate novel structures [Citation66]. In POTM chemistry sulfite (SO32–) was used early on to generate one of the first three-dimensional POTMs which did not use POM fragments [Citation44]. The structure uses six [MoV2O2(µ-S)2]2+ and sixteen SO32–, 12 µ- which connect through two oxygens to equatorial positions, and 4 µ3- which connect through three oxygens to axial positions. This gives an overall tetrahedral Td symmetry. Each µ3-SO3 lies at the one corner of the tetrahedron, with [MoV2O2(µ-S)2]2+ forming the edges of the tetrahedron.
Interestingly, the selenite (SeO32–) anion demonstrates different behavior (). At low (1:1) concentrations selenite forms pairs with [MoV2O2(µ-S)2]2+ attaching to both its axial positions through two of its oxygens in a manner similar to acetate in the Keplerate structures. These oxygen atoms are also able to coordinate to another dimer-selenite pair in its equatorial position, whilst selenite of the second pair likewise coordinates to the equatorial position of the first dimer. This leaves one free equatorial site on both dimers, which is occupied by hydroxide to make a complete bridge. In this way a ring is formed, but rather than uniformly side-on dimer units, with selenites they alternate pointing up and down to give a {Mo16(SeO3)8} ring. In the presence of increased pH, however, hydroxides substitute half the selenites. This forms an unclosed ring made of seven dimer units and three selenite anions, four dimers point outwards as in typical POTM rings, whilst three point upwards, the central dimer and the two terminal dimers. The ring, however, does not perfectly close, instead one of the two terminal dimers becomes part of a triangular group [(MoV2O2(µ-S)2)3(µ3-OH)]5+, a motif that was also seen in the “inverted Keplerate” cube described previously, with the other end of the ring connecting to the far corner of the triangle. This forms a wholly asymmetric unit, essentially consisting of an Mo12 “loop” connected to an Mo6 triangle. Two of these units are fused by the triangular units via a pair of µ3-O bridges on the uncoordinated tips, and also by a singular acetate ligand. This gives an overall {Mo36(SeO3)6} structure with overall C2 symmetry [Citation67].
Figure 9. Reaction coordinates of the selenite POTM system. Structures are positioned according to relative relationship between pH and Se:Mo ratio required for formation. The position does not reflect exact pH or Se:Mo values. A) {MoV27(SeO3)17}, B) {MoV16(SeO3)20}, C) {MoV16(SeO3)} and D) {MoV36(SeO3)6}. Mo, teal; O, red; S, yellow; C, white; Se, purple; coordination positions shown as translucent grey. Red arrows indicate structural relationship and do not necessarily indicate reaction pathway.
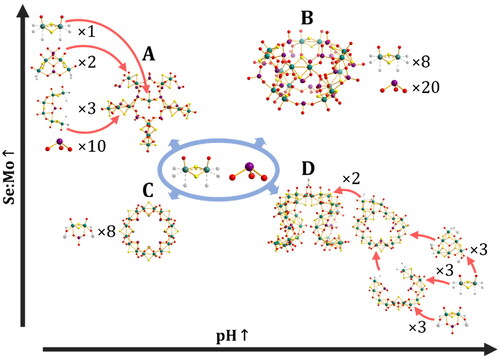
In addition to increasing the concentration of hydroxide (by increasing the pH), new structures may be formed by increasing the concentration of selenite to favor more “selenite dense” structures. Two structures are formed in the presence of excess selenite depending on pH and the degree of excess. At pH 5 the “exo” form {Mo28(SeO3)17} and at even higher concentrations of selenite and pH 8 the “endo” {Mo16(SeO3)20} are formed. The exo form is based on two different ring fragments which may be seen as analogous to the square and diamond coordination modes of squarate. In one a single selenite coordinates to both axial faces of two dimers, whilst another selenite replaces one of the hydroxide bridges between the dimers (diamond-like), in the other a selenite coordinates to both axial faces of a single dimer whilst adjacent dimers coordinate through a single axial position (square-like). Two of the diamond-like building blocks and three of the square-like are rotated so that the plane of the half ring lies perpendicular to the overall star shape and are connected via additional selenite units to each other forming two parallel planes with a central cavity. Finally, a single dimer sits inside the star, coordinated to the connecting selenites. The endo form does not feature any hydroxide bridges or partial POTM rings, and instead consists entirely of µ-selenites and non-bridging selenites connecting individual dimer units. The coordination modes are similar to that of {Mo12(SO3)16}, but lacks any µ3-selenites, forming a molecular cage of overall lower C4V symmetry. The “exo” structure is particularly noteworthy for possessing similar second order building blocks to the squarate system.
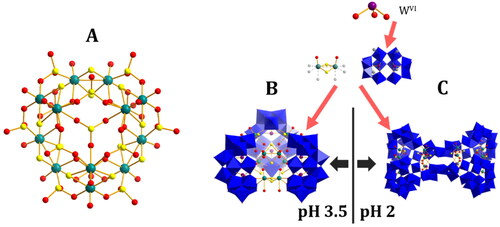
Selenite could also be used to generate in-situ POM fragments to be joined by POTM dimers. Cadot et al. found a system which at pH 3.5 formed a “typical” triangular cluster consisting of three tetra-lacunary tungstate Dawson-type POMs connected by two planes of three POTM dimers each. At lower pH though this extended to a tetrameric structure, with a large separation between the two pairs of Dawson structures, linked by smaller tungstate fragments {WO3(H2O)}, {W2O5} and {W4O12} which are then held together by further bridging POTM dimers (). This cluster holds two differently sized internal cavities. A smaller one in the center and two larger ones next to each pair of Dawson POMs. When crystallized with Na+ the smaller cavity is occupied by the cation, whilst when crystallized with Rb+ it is the larger sites that are occupied [Citation68].
Figure 10. Structures mentioned in this section that do not belong to a specific system of building blocks. A) {MoV12(SO3)16}, B) {MoV12(WVI14Se2O52)3}, C) {MoV16(WVI14Se2O52)5WVI22}. Mo, teal; W, blue polyhedra; O, red; S, yellow; C, white; Se, purple; coordination positions shown as translucent grey.
The tellurite anion (TeO32-–) was found to template a diverse range of POTM structures, distinct from those of selenite (). In addition to obvious changes in atomic radii tellurite has a greater ability to coordinate to additional oxygens to adopt higher coordination numbers forming small tellurite aggregates (examples include Te2O6 and Te3O10 found in the structures following) and square pyramidal (TeO3OH2)2– (this is distinct from the tetrahedral tellurate TeO42– which is in a different oxidation state).
Figure 11. Combination of [MoV2O2(µ-S)2]2+ and tellurite, with (bottom) and without (top) squarate. Colored circles show the building blocks that join to give the structures. A) {MoV8(TeO3)}, B) {MoV24(TeO3)}, C) {MoV20(TeO3)(Te2(µ-O)2O4)(Te2(µ-O)O5)}, D) {MoV24(C4O4)4(TeO4)2}, E) {MoV30(C4O4)3(TeO4)3}, F) {MoV28(C4O4)4(TeO4)2} and G) {MoV72(C4O4)8(Te3O10)4}. Mo, teal; O, red; S, yellow; C, white; Te, brown; Coordination positions shown as translucent grey. Red arrows indicate structural relationship and do not necessarily indicate reaction pathway.
![Figure 11. Combination of [MoV2O2(µ-S)2]2+ and tellurite, with (bottom) and without (top) squarate. Colored circles show the building blocks that join to give the structures. A) {MoV8(TeO3)}, B) {MoV24(TeO3)}, C) {MoV20(TeO3)(Te2(µ-O)2O4)(Te2(µ-O)O5)}, D) {MoV24(C4O4)4(TeO4)2}, E) {MoV30(C4O4)3(TeO4)3}, F) {MoV28(C4O4)4(TeO4)2} and G) {MoV72(C4O4)8(Te3O10)4}. Mo, teal; O, red; S, yellow; C, white; Te, brown; Coordination positions shown as translucent grey. Red arrows indicate structural relationship and do not necessarily indicate reaction pathway.](/cms/asset/d3dfcdb6-c7a3-4ada-b880-340c3120bd8f/gcoo_a_2086049_f0011_c.jpg)
In its simplest form a closed {Mo8(TeO3)} ring is formed, but TeO32– only directly coordinates to one side of this structure. It follows that by removing the “untemplated” half of the ring a highly stable half-ring building block could be formed which could combine in a variety of ways. Pairs of dimers connected to one tellurite formed a half ring which could be connected end-to-end to give a hexameric {Mo24(TeO3)} cluster made of alternating up and down half rings to give an overall “giant chair” structure. Two templating tellurites coordinate to an additional water outside of the ring. The rings connect with each tellurite bridging a single oxygen across the rings, along with an additional bridging hydroxide. A second structure consisting of three different half ring second order building blocks could alternatively be formed. It is made up from one {Mo6(TeO3)}, one {Mo6(Te2(µ-O)2O4)} and one {Mo6(Te2(µ-O)O5}, which fuse together at their open ends to give a {Mo20(TeO3)(Te2(µ-O)2O4)(Te2(µ-O)O5)} structure which is completely asymmetric and chiral [Citation69].
When tellurites are combined with squarate templates a novel building block is formed. The inability of squarate to form {Mo8(C4O4)} rings due to squarate being slightly too large is a major driving factor to its ability to form coordinatively unsaturated second order building blocks. Tellurite in its square pyramidal form is well sized however to replace hydroxide bridges between two dimers to expand the ring enough to fit well around a squarate template. This creates a {Mo8(C4O4)(TeO4)} building block in which the tellurite creates a pair of bidentate binding positions above and below the ring that are not extant when hydroxide is bridging. This can then combine with other fragments to form extended clusters. Two {Mo8(C4O4)(TeO4)} units can combine at right angles with two square-type squarate building blocks to give a {Mo28(C4O4)4(TeO4)2} cluster. They can alternatively combine with the {Mo6(µ3-O)} triangular group attaching at the corners to give a trimeric triskelion structure {Mo30(C4O4)3(TeO4)3}. The partial ring formed around squarate need not be bridged by exactly one tellurite. A pair of tellurites, one lying above, one below the ring may each bridge between one pair of equatorial positions to give {Mo8(C4O4)(TeO3)2}. This leaves the fragment holding two tellurites with uncoordinated oxygen atoms, which can then link into chains of tellurites, or coordinate elsewhere. In the {Mo72(C4O4)8(Te3O10)4} structure four {Te3O10} chains are formed, each coordinates at one terminus to a diamond-type squarate building block. Along the chain two pendant {Mo8(C4O4)} lie, each coordinated to four of the oxygen atoms from the chain. At the other terminus a single oxygen replaces a hydroxide on a {Mo8(C4O4)} attached to a different chain. These four chains lie approximately parallel to each other, two descending from a diamond-type building block at the top and the other two rising from a second diamond-type building block at the bottom of the structure, this time rotated 90° around the central axis of the structure. A total of eight {Mo8(C4O4)} are present. Lastly a pair of square pyramidal tellurites opposite one another may connect four square-type building blocks in a paddlewheel arrangement. Each tellurite coordinates to a single MoV with a pair of oxygens, with each adjacent pair of oxygens coordinating to a different building block for a total of four building blocks. It is the ability of tellurite to increase its coordination number, taking on square pyramidal forms or aggregating that makes these structures possible, thereby making these coordination modes unique to tellurite compared to sulfite and selenite. Of note when combined with squarate, tellurite acts exclusively as a linker, not as a template (coordinating to the axial position), whilst squarate acts as a template.
4. Catalysis
As a field still in its infancy few applications for POTMs have been developed, however, based on their similarity to bulk molybdenum sulfide, POTMs have been explored for use as electrocatalysts in the hydrogen evolution reaction (HER), an important component towards the use of hydrogen as a fuel. Molybdenum sulfide (MoS2) finds applications in industrial hydrodesulfurization (the removal of sulfur from organic compounds by reacting with hydrogen) [Citation1] and has been extensively researched as a candidate electrocatalyst for the HER, of particular interest due to molybdenum’s high natural abundance (compared to noble metals such as platinum). As a molecular form of molybdenum sulfide POTMs have attracted some attention as HER electrocatalysts (). Recent theoretical work [Citation8, Citation70], backed up by experimental observations [Citation3], have indicated that it is exclusively the edge sites that act as active catalysts in molybdenum disulfide. Since molybdenum disulfide tends to form thin sheets with little edge surface area the use of molecular forms presents a potential catalyst in which a much higher percentage (as much as 100%) of the molybdenum sites can act as active sites.
Figure 12. POTM structures that have been used as HER electrocatalysts. A) [MoV2O2(µ-S)2(S2)2]2-, [MoV2O2(µ-S)2(S2)(S4)]2-, [WV2O2(µ-S)2(S2)(S4)]2-, B) {MoV8Ox}, {MoV12BTC}, C) {MoV20PyD2}, {Mo12Nap}, D) {MoV8S8Ale4}, E) {MoV18(MoIV3S4(nta)3)}, {MoV12(Ni(DTO)2)}, F) {Mo12TMT}, no crystal structure exists for {Mo12DFMT} (Ox = oxalate, BTC = benzenetricarboxylate, PyD = 3,5-pyridinedicarboxylate, Nap = 1,4-napthalenedicarboxylate, Ale = alendronic acid, nta = nitrilotriacetate, DTO = dithiooxalate, TMT = tetramethylterephthalate, DFMT = di-trifluoromethylterephthalate). Mo, teal; N, blue; O, red; S, yellow; C, white; Rb, green; W, dark blue; hydrogen omitted for clarity.
![Figure 12. POTM structures that have been used as HER electrocatalysts. A) [MoV2O2(µ-S)2(S2)2]2-, [MoV2O2(µ-S)2(S2)(S4)]2-, [WV2O2(µ-S)2(S2)(S4)]2-, B) {MoV8Ox}, {MoV12BTC}, C) {MoV20PyD2}, {Mo12Nap}, D) {MoV8S8Ale4}, E) {MoV18(MoIV3S4(nta)3)}, {MoV12(Ni(DTO)2)}, F) {Mo12TMT}, no crystal structure exists for {Mo12DFMT} (Ox = oxalate, BTC = benzenetricarboxylate, PyD = 3,5-pyridinedicarboxylate, Nap = 1,4-napthalenedicarboxylate, Ale = alendronic acid, nta = nitrilotriacetate, DTO = dithiooxalate, TMT = tetramethylterephthalate, DFMT = di-trifluoromethylterephthalate). Mo, teal; N, blue; O, red; S, yellow; C, white; Rb, green; W, dark blue; hydrogen omitted for clarity.](/cms/asset/f5a46500-c85c-4a22-8c33-fcc2291b5f82/gcoo_a_2086049_f0012_c.jpg)
The high activity observed from amorphous forms of molybdenum sulfide [Citation4, Citation5] further implies the effectiveness of POTMs as HER electrocatalysts. The structure of amorphous molybdenum sulfide is highly reminiscent of that of many POTMs, featuring distinct [MoV(µ3-S)(µ-S2)3]7+ blocks which link via terminal disulfides coordinating through MoV, and where not bridging, form MoV=O bonds [Citation71]. It is these terminal oxo groups that are thought to be crucial for the activity, being lost and forming the active sites during catalysis. The presence of MoV and MoV=O bonds are different from crystalline molybdenum disulfide but are also present in the POTMs described here. Based on these observations POTMs seem ideal candidates for HER electrocatalysis.
Early work showed a molecular molybdenum sulfide dimer, [Cp2MoIV(µ-S)2(η2-µ-SCH2S)], and related derivatives were highly active towards HER, with an overpotential as low as −120 mV, comparable to some of the best MoS2 on graphite catalysts and with efficiency close to 100% [Citation72]. They also note its compatibility with CO which poisons platinum catalysts. More recently precursors to [Mo2O2(µ-S)2]2+ were investigated [Citation73]. Three complexes, [Mo2O2(µ-S)2(η2-S2)(η2-S4)]2–, [Mo2O2(µ-S)2(η2-S2)2]2– and [W2O2(µ-S)2(η2-S2)(η2-S4)]2–, were studied finding that the molybdenum catalysts were able to achieve overpotentials as low as −114 mV whilst the tungsten catalysts performed worse at −227 mV overpotential. Of note these catalysts were stable under cycling conditions in aqueous solution at pH 0. Commercial water-splitting via HER requires aqueous conditions, so a candidate catalyst must be compatible with these conditions, which this class of catalysts are. Theoretical calculations show that, much like the bulk MoS2 these dimers mimic, the mechanism proceeds via protonation of the terminal η2-S2 groups.
The lack of η2-S2 groups in self-condensed POTM rings may then be expected to prevent the activity of them towards HER, however, this has proven not to be the case [Citation74]. Cadot et al. were able to show both {Mo8Ox} and {Mo12BTC} (Ox = oxalate, BTC = benzenetricarboxylate) to be active electrocatalysts for HER. DFT calculations suggested that protonation may occur on the carboxylate groups of the templates, thereby replacing the role of the η2-S2 groups and indicating a synergistic behavior between the ring and the template. The catalysts were investigated in homogenous conditions in DMF. When appropriately strong acids were used, peak potentials of −910 mV (vs. SCE) ({Mo12BTC} in the presence of HClO4) and −1000 mV (vs. SCE) ({Mo8Ox} in the presence of HClO4) were achieved. More recently Zhang et al. showed that their {Mo20(PyD)2} (described earlier) and their {Mo12(Nap)} (PyD = 3,5-pyridinedicarboxylate, Nap = 1,4-napthalenedicarboxylate) rings were active for HER in organic medium [Citation56]. The bisphosphonate-bridged complex {Mo8S8(Ale)4} (also described earlier) showed catalytic activity in acetonitrile, but the oxo form {Mo8O8(Ale)4} in which [Mo2O2(µ-S)2]2+ dimers are replaced with [Mo2O2(µ-O)2]2+ showed no activity. From this it was inferred that the bridging sulfide ligands were crucial to the electrocatalytic performance.
Aqueous electrocatalysis is also possible. Two different clusters incorporating captured metals into the template, {Mo12(NiIIdto2)} [Citation40] and {Mo18(MoIV3S4(nta)3)} [Citation41] (dto = dithiooxalate, nta = nitrilotriacetate), both showed catalytic activity, with uncomplexed [MoIV3S4(nta)3]5– showing no activity without the POTM ring. Rings with purely organic templates were also able to show activity in aqueous medium [Citation51]. A series of terephthalate derivatives were used as templates, along with benzenetricarboxylate by Cadot et al. [Citation51] to compare how changes to the pKa of the template may impact the HER activity. It was found that the less basic di(trifluoromethyl)terephthalate showed less activity than other, more basic templates. Benzenetricarboxylate showed the greatest activity, with an onset potential of −180 mV. Whilst it is similar in basicity to the other investigated template, tetramethylterephthalate, it presents an additional coordinating carboxylate group and would therefore provide a 50% increase in the available sites for a mechanism involving protonation at the ligand. Based on experimental observations, DFT calculations and evidence from other MoS catalysts they propose a mechanism using the carboxylate as a proton relay. The carboxylate is initially protonated breaking the O-Mo bond. The dimer is then reduced at both MoV centers. The proton migrates to the uncoordinated MoIV axial position, forming a MoV hydride. A second proton adds to the now free carboxylate oxygen, and lastly the hydride migrates to this proton, eliminating H2 gas.
The MoIV-based building block {MoIV3S4} is on its own an active electrocatalyst for HER [Citation75]. In solution it can associate with the large toroidal POM {P8W48} in ratios up to 4:1. Cadot et al. demonstrated that when supported in this way the HER activity dramatically increased [Citation76]. This increase was not seen using non-POM ligands to support {MoIV3S4}, and titration of {MoIV3S4} into {P8W48} showed no significant increase in activity beyond a 4:1 ratio (the loading limit). Based on this evidence the two components appear to exhibit a synergistic behavior, wherein the POM can act as a delocalised electron resevoir for the thiometalate catalyst. When immobilized onto a photocathode the catalytic enhancement remained despite the heterogenous conditions and demostrated hydrogen evolution under simulated sunlight reaching a current density of −10 mA/cm−2 at +0.07 V (vs. RHE).
Following this method a dimeric cluster {(MoIV3S4)2(PW11)2} was synthesized which also showed enhanced activity towards electrocatalytic HER [Citation75]. When combined with an Ir-based photosensitizer and a proton/electron donor the complex was able to perform solar-driven hydrogen evolution. The performance was signficantly better compared to similarly structured Ni-based catalysts, and when POM-free {Mo3S4} was used the activity was 25% lower. Further to this the addition of {PW11} actually inhibited the activity of {Mo3S4} by almost 50% when the two components were not connected, attributed to competition for assocaition with the photosensitizer.
Although hydrogen evolution is of great interest for prospects of hydrogen generation as a fuel source, it has yet to be realized commercially. In comparison hydrodesulfurization of petroleum by MoS2 is an economically crucial use of molybdenum sulfide catalysts [Citation77]. The effectiveness as hydrodesulfurization catalysts of three different POTM rings, {Mo10}, {Mo12BDC} and {Mo12BTC} (BDC = 1,4 bezenedicarboxylate), was investigated by supporting them on the mesoporous silica substrate SBA-15 [Citation78]. All three were effective at desulfurization of thiophene with essentially complete conversion after 20 h at 320 °C. The {Mo4(Tart)2} (Tart = tartarate) ring (described earlier) could also be immobilized on SBA-15 and used for dehydrosulfurization of thiophene in a similar manner [Citation55].
Cadot et al. investigated the electrochemical properties of POTMs formed by adding sulfide dimers ([MV2O2(µ-S)2]2+) (M = Mo or W) to the lacunary [SiWVI10O36]8– Keggin [Citation79] and found that they were active for electrochemical reduction of iodate [Citation80]. Compared to the non-thio form [SiWVI12O40]4–, [SiWVI10O36(WV2O2(µ-S)2]6– showed an almost ten-fold increase in activity.
5. Conclusion
Over the past ten years the field of POTMs has expanded in many unexpected ways. Each new structure demonstrates the effective utilization of the newly designed building blocks that can be combined in different ways. The diversity of coordination modes and structural motifs combined with a template-driven self-assembly approach gives a functionally unlimited range of plausible structures which may be envisioned based on combining distinct building blocks and template moieties. A representative example is the case of the squarate anion which can template the formation of constituents and induce appropriate curvature but is disinclined to form small clusters with just itself as a template, has proved to be highly beneficial for the construction of high nuclearity, geometrically complex POTMs. The main obstacle that remains is the monumental task of exploring the vast chemical space that is generated by these dynamic, multicomponent systems. In addition to the use of high-throughput automation techniques, diligent mapping of the reaction conditions giving rise to distinct building block fragments will help to drive targeted exploration towards specific desired outcomes. The screening of small templates that can generate secondary building blocks could pave the way for further expansion of this family of clusters and discovery of new functionalities.
POTMs have been shown to perform comparably to the best noble-metal free electrocatalysts for HER. It should be kept in mind that the performance of bulk material catalysts such as MoS2 are the product of many years of tuning in terms of processing techniques and doping, which POTMs are yet lacking. There is every reason to believe that focused research could yield even better performing POTM catalysts for HER. Promising results suggest a synergistic effect between Mo-S cores which may act as catalyst and supporting POMs which can act as proton/electron reservoirs. Early work suggests that POTMs may also be effective hydrodesulfurization catalysts which is of industrial importance. The development of larger structures that may act as a host of a variety of smaller molecules with complex morphology, increased porosity and surface areas opens new horizons for performing efficiently catalyzed reactions in confined spaces.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- R.R. Chianelli, M. Daage, M.J. Ledoux. Adv. Catal., 40, 177 (1994).
- C.D. Garner. The Chemical Nature of the Molybdenum Centres in Enzymes, Vol. 19, Elsevier B.V. (1994).
- T.F. Jaramillo, K.P. Jorgensen, J. Bonde, J.H. Nielsen, S. Horch, I. Chorkendorff. Science, 317, 100 (2007).
- D. Merki, S. Fierro, H. Vrubel, X. Hu. Chem. Sci., 2, 1262 (2011).
- C.G. Morales-Guio, X. Hu. Acc. Chem. Res., 47, 2671 (2014).
- T. Sasaki, T. Suzuki. Appl. Catal. A: Gen., 484, 79 (2014). 83
- X. Zou, Y. Zhang. Chem. Soc. Rev., 44, 5148 (2015).
- B. Hinnemann, P.G. Moses, J. Bonde, K.P. Jørgensen, J.H. Nielsen, S. Horch, I. Chorkendorff, J.K. Nørskov. J. Am. Chem. Soc., 127, 5308 (2005).
- M.T. Pope, A. Müller. Angew. Chem. Int. Ed. Engl., 30, 34 (1991).
- H.N. Miras, J. Yan, D.L. Long, L. Cronin. Chem. Soc. Rev., 41, 7403 (2012).
- U. Kortz, A. Müller, J. van Slageren, J. Schnack, N.S. Dalal, M. Dressel. Coord. Chem. Rev., 253, 2315 (2009).
- E. Cadot, V. Béreau, F. Sécheresse. Inorg. Chim. Acta, 239, 39 (1995).
- W.G. Klemperer, C. Schwartz. Inorg. Chem., 24, 4459 (1985).
- D. Coucouvanis, A. Toupadakis, A. Hadjikyriacou. Inorg. Chem., 27, 3272 (1988).
- W. Rittner, A. Müller, A. Neumann, W. Bäther, R.C. Sharma. Angew. Chem. Int. Ed. Engl., 18, 530 (1979).
- V. Béreau, E. Cadot, H. Bögge, A. Müller, F. Sécheresse. Inorg. Chem., 38, 5803 (1999).
- S.A. Cohen, E.I. Stiefel. Inorg. Chem., 24, 4657 (1985).
- F.A. Cotton, Z. Dori, R. Llusar, W. Schwotzer. J. Am. Chem. Soc., 107, 6734 (1985).
- A. Müller, S. Sarkar, R.G. Bhattacharyya, S. Pohl, M. Dartmann. Angew. Chem. Int. Ed. Engl., 17, 535 (1978).
- D.T. Richens, P.A. Pittet, A.E. Merbach, M. Humanes, G.J. Lamprecht, B.L. Ooi, A.G. Sykes. Dalton Trans., 2305 (1993).
- E. Cadot, B. Salignac, S. Halut, F. Sécheresse. Angew. Chem. Int. Ed., 37, 611 (1998).
- H. Akashi, T. Shibahara, H. Kuroya. Polyhedron, 9, 1671 (1990).
- E. Cadot, V. Béreau, F. Sécheresse. Inorg. Chim. Acta, 252, 101 (1996).
- J. Marrot, M.A. Pilette, M. Haouas, S. Floquet, F. Taulelle, X. López, J.M. Poblet, E. Cadot. J. Am. Chem. Soc., 134, 1724 (2012).
- R. Hernandez-Molina, M.N. Sokolov, A.G. Sykes. Acc. Chem. Res., 34, 223 (2001).
- E. Cadot, B. Salignac, J. Marrot, A. Dolbecq, F. Sécheresse. Chem. Commun., 10, 261 (2000).
- B. Salignac, S. Riedel, A. Dolbecq, F. Sécheresse, E. Cadot. J. Am. Chem. Soc., 122, 10381 (2000).
- E. Cadot, J. Marrot, F. Sécheresse. Angew. Chem., 113, 796 (2001).
- E. Cadot, F. Sécheresse. Chem. Commun., 2, 2189 (2002).
- S. Floquet, J. Marrot, E. Cadot. CR Chim., 8, 1067 (2005).
- J.-F. Lemonnier, S. Floquet, J. Marrot, E. Terazzi, C. Piguet, P. Lesot, A. Pinto, E. Cadot. Chemistry, 13, 3548 (2007).
- J.-F. Lemonnier, S. Floquet, A. Kachmar, M.-M. Rohmer, M. Bénard, J. Marrot, E. Terazzi, C. Piguet, E. Cadot. Dalton Trans., 3043, 3043 (2007).
- J.-F. Lemonnier, A. Kachmar, S. Floquet, J. Marrot, M.-M. Rohmer, M. Bénard, E. Cadot. Dalton Trans., 4565 (2008).
- A. Kachmar, S. Floquet, J.F. Lemonnier, E. Cadot, M.M. Rohmer, M. Bénard. Inorg Chem., 48, 6852 (2009).
- A. Dolbecq, E. Cadot, F. Sécheresse. Chem. Commun., 12, 2293 (1998).
- E. Cadot, A. Dolbecq, B. Salignac, F. Sécheresse. Chem. Eur. J., 5, 2396 (1999).
- E. Cadot, B. Salignac, T. Loiseau, A. Dolbecq, F. Sécheresse. Chem. Eur. J., 5, 3390 (1999).
- A. Dolbecq, C. du Peloux, A.-L. Auberty, S.A. Mason, P. Barboux, J. Marrot, E. Cadot, F. Sécheresse. Chem. Eur. J., 8, 349 (2002).
- E. Cadot, M.-J. Pouet, C. Robert-Labarre, C. du Peloux, J. Marrot, F. Sécheresse. J. Am. Chem. Soc., 126, 9127 (2004).
- A. Hijazi, J.C. Kemmegne-Mbouguen, S. Floquet, J. Marrot, C.R. Mayer, V. Artero, E. Cadot. Inorg. Chem., 50, 9031 (2011).
- S. Duval, S. Floquet, C. Simonnet-Jégat, J. Marrot, R.N. Biboum, B. Keita, L. Nadjo, M. Haouas, F. Taulelle, E. Cadot. J. Am. Chem. Soc., 132, 2069 (2010).
- J.-F. Lemonnier, S. Duval, S. Floquet, E. Cadot. Isr. J. Chem., 51, 290 (2011).
- E. Cadot, M.N. Sokolov, V.P. Fedin, C. Simonnet-Jégat, S. Floquet, F. Sécheresse. Chem. Soc. Rev., 41, 7335 (2012).
- H.N. Miras, J.D. Woollins, A.M. Slawin, R. Raptis, P. Baran, T.A. Kabanos. Dalton Trans., 6, 3668 (2003).
- V.S. Korenev, S. Floquet, J. Marrot, M. Haouas, I.-M. Mbomekallé, F. Taulelle, M.N. Sokolov, V.P. Fedin, E. Cadot. Inorg. Chem., 51, 2349 (2012).
- C. du Peloux, A. Dolbecq, F. Sécheresse. Inorg. Chem. Commun., 5, 333 (2002).
- S. Floquet, S. Draoui, J. Marrot, F. Millange, M. Frigoli, E. Cadot. J. Clust. Sci., 25, 811 (2014).
- H. Zang, H.N. Miras, J. Yan, D.-L. Long, L. Cronin. J. Am. Chem. Soc., 134, 11376 (2012).
- H. Zang, A. Surman, D. Long, L. Cronin, H.N. Miras. Chem. Commun. (Camb), 52, 9109 (2016).
- D.S. Ogienko, A.I. Smolentsev, S.N. Konchenko. J. Struct. Chem., 56, 762 (2015).
- A. Hijazi, J.C. Kemmegne-Mbouguen, S. Floquet, J. Marrot, J. Fize, V. Artero, O. David, E. Magnier, B. Pégot, E. Cadot. Dalton Trans., 42, 4848 (2013).
- H.-Y. Zang, A.R. de la Oliva, H.N. Miras, D.-L. Long, R.T. McBurney, L. Cronin. Nat. Commun., 5, 3715 (2014).
- H.E. Moll, J.C. Kemmegne-Mbouguen, M. Haouas, F. Taulelle, J. Marrot, E. Cadot, P. Mialane, S. Floquet, A. Dolbecq. Dalton Trans., 41, 9955 (2012).
- J.-D. Compain, P. Mialane, J. Marrot, F. Sécheresse, W. Zhu, E. Oldfield, A. Dolbecq. Chemistry, 16, 13741 (2010).
- T.T. Qian, Y.J. Cui, Z. Xin, A.Q. Jia, Q.F. Zhang. Z. Naturforsch. - Sect. B J. Chem. Sci., 72, 475 (2017).
- Q.-Y. Xu, Z. Xin, M.-J. Hu, H.-T. Shi, A.-Q. Jia, Q.-F. Zhang. J. Cluster Sci., 4, (2022). https://link.springer.com/article/10.1007/s10876-022-02219-4#citeas
- V.S. Korenev, A.G. Boulay, A. Dolbecq, M.N. Sokolov, A. Hijazi, S. Floquet, V.P. Fedin, E. Cadot. Inorg. Chem., 49, 9740 (2010).
- A. Müller, E. Krickemeyer, H. Bögge, M. Schmidtmann, F. Peters. Angew. Chem. Int. Ed., 37, 3359 (1998).
- C. Schäffer, A.M. Todea, H. Bögge, E. Cadot, P. Gouzerh, S. Kopilevich, I.A. Weinstock, A. Müller. Angew. Chem. Int. Ed. Engl., 50, 12326 (2011).
- F. Bannani, S. Floquet, N. Leclerc-Laronze, M. Haouas, F. Taulelle, J. Marrot, P. Kögerler, E. Cadot. J. Am. Chem. Soc., 134, 19342 (2012).
- V.S. Korenev, A.G. Boulay, M. Haouas, F. Bannani, V.P. Fedin, M.N. Sokolov, E. Terazzi, S. Garai, A. Müller, F. Taulelle, J. Marrot, N. Leclerc, S. Floquet, E. Cadot. Chemistry, 20, 3097 (2014).
- A. Müller, E. Krickemeyer, S. Dillinger, H. Bögge, W. Plass, A. Proust, L. Dloczik, C. Menke, J. Meyer, R. Rohlfing. Z. Anorg. Allg. Chem., 620, 599 (1994).
- B. Krebs, I. Paulat-Böschen. Acta Crystallogr. B Struct. Crystallogr. Cryst. Chem., 38, 1710 (1982).
- C.J. Richmond, H.N. Miras, A.R. De La Oliva, H. Zang, V. Sans, L. Paramonov, C. Makatsoris, R. Inglis, E.K. Brechin, D.-L. Long, L. Cronin. Nat. Chem., 4, 1037 (2012).
- H.-Y. Zang, H.N. Miras, D.-L. Long, B. Rausch, L. Cronin. Angew. Chem. Int. Ed. Engl., 52, 6903 (2013).
- H.N. Miras, G.I. Chilas, L. Cronin, T.A. Kabanos. Eur. J. Inorg. Chem., 2013, 1620 (2013).
- H.-Y. Zang, J.-J. Chen, D.-L. Long, L. Cronin, H.N. Miras. Adv. Mater., 25, 6245 (2013).
- S. Floquet, P.A. Abramov, E. Cadot. Polyhedron, 175, 114233 (2020).
- J.W. Purcell, D.-L. Long, E.C. Lee, L. Cronin, H.N. Miras. Dalton Trans., 47, 6283 (2018).
- P. Raybaud, J. Hafner, G. Kresse, S. Kasztelan, H. Toulhoat. J. Catal., 189, 129 (2000).
- P.D. Tran, T.V. Tran, M. Orio, S. Torelli, Q.D. Truong, K. Nayuki, Y. Sasaki, S.Y. Chiam, R. Yi, I. Honma, J. Barber, V. Artero. Nat. Mater., 15, 640 (2016).
- A.M. Appel, D.L. DuBois, M. Rakowski DuBois. J. Am. Chem. Soc., 127, 12717 (2005).
- J. McAllister, N.A.G. Bandeira, J.C. McGlynn, A.Y. Ganin, Y.-F. Song, C. Bo, H.N. Miras. Nat. Commun., 10, 370 (2019).
- B. Keita, S. Floquet, J.-F. Lemonnier, E. Cadot, A. Kachmar, M. Bénard, M.-M. Rohmer, L. Nadjo. J. Phys. Chem. C, 112, 1109 (2008).
- J. Al Cheikh, R. Zakari, A.C. Bhosale, A. Villagra, N. Leclerc, S. Floquet, P.C. Ghosh, A. Ranjbari, E. Cadot, P. Millet, L. Assaud. Mater. Adv., 1, 430 (2020).
- J. Tourneur, B. Fabre, G. Loget, A. Vacher, C. Mériadec, S. Ababou-Girard, F. Gouttefangeas, L. Joanny, E. Cadot, M. Haouas, N. Leclerc-Laronze, C. Falaise, E. Guillon. J. Am. Chem. Soc., 141, 11954 (2019).
- M. Li, H. Li, F. Jiang, Y. Chu, H. Nie. Catal. Today, 149, 35 (2010).
- Z. Xin, W. Wei, M. Chen, A.Q. Jia, Q.F. Zhang. Catal. Commun., 64, 86 (2015). 90
- J.C. Kemmegne-Mbouguen, S. Floquet, D. Zang, A. Bonnefont, L. Ruhlmann, C. Simonnet-Jégat, X. López, M. Haouas, E. Cadot. New J. Chem., 43, 1146 (2019).
- J.C. Kemmegne-Mbouguen, S. Floquet, E. Cadot. CR Chim., 24, 91 (2021).