
Abstract
A polymerase chain reaction (PCR) assay was developed that amplified a 170-bp fragment of the intergenic spacer region of Sclerotinia sclerotiorum, the cause of white mould. Sensitivity was 10 S. sclerotiorum ascospores per DNA extraction (0.2 ascospores per PCR reaction). The presence of soil did not affect sensitivity at 50, 100 and 500 ascospores/DNA extraction, but reduced sensitivity at 25 and 10 ascospores/DNA extraction by 10% and 30%, respectively. The assay did not amplify DNA of Botrytis cinerea but detected S. minor and S. trifoliorum. Utility of the test for detection of S. sclerotiorum ascospores in bean fields was demonstrated using rotating impaction samplers over two seasons. The use of the test in combination with an impaction sampler may provide benefits in time, sensitivity and specificity compared with visual identification and enumeration of spores from traps only. This system may provide an opportunity to schedule fungicides during periods of inoculum presence for disease management.
Introduction
White mould caused by the fungus Sclerotinia sclerotiorum is one of the most economically damaging diseases of bean (Phaseolus vulgaris L.) and many other crops (Boland & Hall Citation1987). Management of white mould is challenging due to its persistence in soil as sclerotia and its wide host range. In bean, infection of the senescing flowers occurs from windborne ascospores produced following carpogenic germination of sclerotia (Williams & Western Citation1965; Schwartz & Steadman Citation1978). Infected senescing flowers may fall and adhere to other plant parts, and act as a nutrient base for the fungus to infect pods, leaves and stems (Sutton & Deverall Citation1983). Yield loss in bean fields occurs through rotting of stems leading to pod abscission, and smaller and diseased pods (Boland & Hall Citation1987). In Tasmania, Australia, if more than 5% of pods are affected by white mould then loads may be rejected by the processor.
Routine management of white mould in bean is through prophylactic protectant fungicide usage. Disease forecasting systems based upon inoculum presence have been developed for a range of pathosystems to aid management decisions, including diseases caused by Sclerotinia spp. (e.g. Clarkson et al. Citation2004; McDonald & Boland Citation2004). Various approaches have been used to quantify the presence of Sclerotinia spp. inoculum, including direct observation of apothecia and their frequency (McCartney & Lacey Citation1991, Citation1999; Steadman & O’Keefe Citation1999; Clarkson et al. Citation2004). Other studies have incorporated additional risk factors such as host susceptibility and canopy characteristics (Turkington & Morall Citation1993) and environmental conditions (e.g. Gugel & Morall Citation1986; Young et al. Citation2005; Harikrishnan & del Río Citation2008).
A common element among many of these forecasting systems is an accurate and precise quantification of ascosporic inoculum. Semi-selective media have been used to detect ascospores directly but their practical deployment in commercial fields has been limited (Ben-Yephet & Bitton Citation1985). Traditionally, this has been conducted by the visual enumeration on various types of spore traps (Hartill Citation1980) and the derivation of statistical relationships between spore density and disease prevalence and incidence (McCartney & Lacey Citation1991, Citation1999). However, S. sclerotiorum ascospores are nondescript and often difficult to differentiate from other fungi on the basis of morphology alone. Microscopic examination of the tapes from spore traps is also time consuming and laborious, and may limit the practicality of detection systems over large production regions based on the presence of inoculum. For example, Rogers et al. (Citation2009) found that it was not possible to detect fewer than 30 to 40 ascospores per day on Burkard traps unless the entire tape was examined and this was deemed unfeasible. The potential masking of hyaline spores, including S. sclerotiorum ascospores, by soil and debris, or high concentrations of pigmented fungal fragments, may also distort results (Rogers et al. Citation2009).
Molecular techniques have now been used in a range of pathosystems to monitor the inoculum presence and to initiate fungicide applications (e.g. Falacy et al. Citation2007; Carisse et al. Citation2009; Gent et al. Citation2009; Klosterman et al. Citation2014). Similar approaches have been attempted for S. sclerotiorum (Freeman et al. Citation2002; Rogers et al. Citation2009). Freeman et al. (Citation2002) developed polymerase chain reaction (PCR) primers and methodologies for the detection of S. sclerotiorum ascospores on adhesive tape from Burkard volumetric spore traps and infected petals. However, in some crops and pathosystems, the technique may be limited by the inability of the primers, designed within the internal transcribed spacer region, to differentiate the closely related species, Botrytis cinerea (Yin et al. Citation2009; Qin et al. Citation2010). The objective of this study was to develop a sensitive and specific PCR assay for the detection of S. sclerotiorum.
Materials and methods
PCR development
Sclerotinia sclerotiorum ascospores were obtained from M. Boosalis, University of Nebraska, Lincoln, USA. Varying concentrations of ascospores (100, 250, 500, 1000, and 5000 mL−1) were obtained after dilution in sterilised distilled water. DNA was extracted from ascospore suspensions with a PowerSoil® DNA Isolation Kit (MO BIO Laboratories Inc, Carlsbad, CA, USA) according to the manufacturer’s protocol. The volume of ascospore suspension used in each extraction was 100 μL for each of the final concentrations of 10, 25, 50, 100 and 500 ascospores/extraction. Ten replicate extractions were conducted for each concentration (alone and with 10 mg of ferrosol soil typically found in bean fields in Tasmania, Australia). For negative controls, 10 DNA extractions were conducted from 100 μL dH2O (with and without soil).
Forward and reverse primers were designed based on sequence data from the intergenic spacer (IGS) region (S. sclerotiorum; GenBank Accession AF342328). The sequence was aligned using Vector NTI Suite v10.0.1 (Life Technologies, Carlsbad, CA, USA). Potential primer sites were identified with Invitrogen online software (OligoPerfect™ Designer) and analysed with Amplify3 version 3.1.4 (http://engels.genetics.wisc.edu/amplify/). Two primer sites, SIG2598 (5′-ATCAGGGTGGTCCAGTTTTG-3′) and SIG2767 (5′-TCGCATTCATAGAACGCTTG-3′), were selected and predicted to amplify a 170-bp product.
PCR conditions were optimised on a gradient thermocycler (Bio-Rad C-1000, Bio-Rad Laboratories Pty Ltd, Gladesville, NSW, Australia). Reactions were performed in a total volume of 20 μL. The PCR reaction mixture consisted of 1 × PCR buffer with 1.5 mM MgCl2 (Applied Biosystems Inc, Foster City, CA, USA), 200 μM dNTPs, 0.2 μM each primer, 0.5 units Taq polymerase and DNA (20–40 ng). Optimised PCR reactions were performed on a GeneAmp PCR Systems 2400 thermocycler (Perkin Elmer, Norwalk, CT, USA). Cycling conditions were an initial denaturation for 10 min at 94 °C, followed by 40 cycles of 30 s denaturation at 94 °C, 30 s annealing at 60 °C, 1 min extension at 72 °C and a final 10 min extension step at 72 °C. PCR products were visualised on a 1.25% agarose gel post-stained with GelRed (Biotium Inc, Hayward, CA, USA). Product size was determined using a 100-bp ladder.
Specificity was tested with other Sclerotinia spp., B. cinerea and other fungal species (). DNA was extracted from mycelia of these isolates with the DNeasy Plant Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions.
Table 1 Fungal species and isolates used to test specificity of the SIG2598F/SIG2767R primers identified within the IGS region for Sclerotinia sclerotiorum detection.
Detection of Sclerotinia sclerotiorum ascospores on spore traps
The PCR assay was tested on a rotation arm impaction (Rotorod) sampler (Model 20, Sampling Technologies Inc, Minnetonka, MN, USA).
Samplers were placed in eight bean fields in northwestern Tasmania over two seasons (n = 4 in each of 2008–09 and 2009–10). Traps were positioned approximately 40 cm above the ground near the centre of the fields. The spore monitoring period in each field was pre-flowering to 2–9 days before harvest. This corresponded to sampling periods of 29 December 2008 to 18 March 2009, and 12 January to 18 March 2010.
The Rotorod sampler collects spores on two spinning (2400 rpm) rods. Each Rotorod sampler was fitted with pairs of stainless steel rods (50 mm long by 1 mm). Rods were covered with a thin coating of silicone grease and collected at 72 or 96 h intervals with sterile forceps. Sixty microlitres of MO BIO extraction solution S1 and 200 μL of MO BIO solution IRS were added to the tubes. The tubes were then secured horizontally on to a MO BIO Vortex adapter tube holder and mixed at maximum speed for 10 min. Tubes were then placed into boiling water for 2 min before being centrifuged at 10,000 g for 30 s. DNA was extracted according to the manufacturer’s recommendations (PowerSoil® DNA Isolation Kit). Each sample was tested with the PCR protocol described above. Samples that were negative with the initial 1 µL of DNA template were tested a second time with 3 µL.
Disease assessments and agronomic data
In the bean fields where Rotorod samplers were deployed, disease assessments were conducted to relate the incidence of diseased plants and pods to the frequency of ascospore detection by PCR. Disease assessments were conducted using a cluster sampling strategy as described by Jones et al. (Citation2011). In brief, disease assessments were conducted 2–9 days before harvest. Pods and plants at 64 sampling points per field were examined for signs and symptoms of white mould. In each field, four to five transects were established and plants were examined at 12–16 equally spaced points along each transect that spanned the length of each field. At each sampling point, 10 plants and 20 pods within a row were assessed for white mould. The 20 pods were selected arbitrarily from one to two plants, depending on the cultivar and number of pods per plant. Information on fungicide application dates (boscalid 500 g/kg; Filan, BASF, Victoria, Australia) was obtained from growers and the processor.
Data analysis
Linear regression was used to quantify relationships between the percentage of sampling periods where S. sclerotiorum ascospores were detected and the end of season incidence of diseased pods and plants. Regression was also used to quantify if there was a relationship between the frequency of sampling periods where ascospores were detected before the first fungicide application and the incidence of pods and plants with white mould.
Analyses were conducted in SAS version 9.4 (SAS Institute, Cary, North Carolina, USA).
Results
PCR development
Primers designed in this study amplified a predicted 170-bp fragment from isolates of S. sclerotiorum, S. trifoliorum and S. minor but did not amplify other fungal species tested, including B. cinerea ().
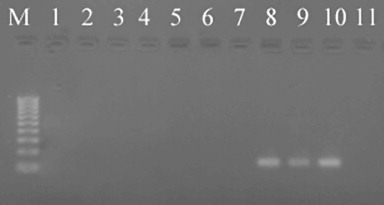
The PCR method detected DNA of S. sclerotiorum as low as 10 spores per DNA extraction (0.2 ascospores per PCR) (). Detection was not affected by the presence of soil for ascospore concentrations of 50, 100 and 500 ascospores per DNA extraction. However, the addition of soil reduced PCR sensitivity (i.e. the number of replicates testing positive) at 25 and 10 ascospores per DNA extraction by 10% and 30%, respectively ().
Table 2 Number of positive PCR tests/total number of replicate samples for each Sclerotinia sclerotiorum ascospore concentration (with and without 10 mg soil).
Detection of Sclerotinia sclerotiorum ascospores on spore traps
Sclerotinia sclerotiorum ascospores were broadly distributed over the sampling period in the fields and detected by PCR on spore traps obtained from all fields sampled. In fields where spore traps were deployed before the first fungicide application, ascospores were detected in all but one field ().
Table 3 Detection of ascosporic inoculum of Sclerotinia species on a rotating arm impaction spore trap by PCR amplification using primers SIG2598F and SIG2767R, and disease incidence of pods and plants in each of eight bean fields in Tasmania, Australia.
There was not an overall association between the percentage of sampling periods when ascospores were detected and diseased pods (P = 0.185) or plants (P = 0.224). Moreover, no significant associations were found between the frequency of sampling periods in which ascospores were detected and the incidence of white mould on pods (P = 0.071) or plants (P = 0.414) ().
Discussion
This study designed primers targeting the IGS region of the genome to provide specificity between Sclerotinia spp. and B. cinerea for ascosporic inoculum of Sclerotinia species. In addition to S. sclerotiorum, these primers were also able to detect S. trifoliorum and S. minor, similar to those of Freeman et al. (Citation2002) and Qin et al. (Citation2010). Sclerotinia minor has the potential to carpogenically germinate and produce viable ascospores in southeastern Australia (Ekins et al. Citation2002). Moreover, carpogenic germination of S. minor has been described in perennial pyrethrum fields in northwest Tasmania and S. minor was isolated at low frequency from pyrethrum flowers affected by Sclerotinia flower blight (O’Malley Citation2012). Isolations from diseased beans over 3 years found S. minor in less than 5% of pods and only in one district (Cressy/Longford); however, carpogenic germination of apothecia was not observed. The contribution of S. minor ascosporic inoculum to white mould epidemics in bean fields in Tasmania is therefore considered to be rare and insignificant compared with the inoculum contribution from S. sclerotiorum. The potential for contribution of ascospores from S. trifoliorum to reduce the utility of this test as a spore-trapping procedure is further unlikely because carpogenic germination of this species is almost exclusively observed in the autumn (Williams & Western Citation1965), when the bean harvest has been completed. Freeman et al. (Citation2002) also proposed that the biology of the Sclerotinia spp. was sufficiently different and that detection of ascospores of species other than S. sclerotiorum in fields of oilseed rape was unlikely. Moreover, Abd-Elmagid et al. (Citation2013) developed species-specific primers to distinguish between the economically important Sclerotinia spp. (S. sclerotiorum, S. minor, S. trifoliorum and S. homoeocarpa). These primers target different genes for each of these species and were found to be highly sensitive and capable of multiplexing for detection and discrimination of species using DNA extracted from mycelia (Abd-Elmagid et al. Citation2013). The use of these primers for ascosporic inoculum detection and species differentiation has not been tested.
The PCR test developed in this study allowed very low amounts (i.e. 50 ascospores per DNA extraction) of airborne S. sclerotiorum inoculum to be detected consistently, even in the presence of potential PCR inhibitors found in soil. This method was therefore more sensitive than the PCR diagnostic methods reported by Freeman et al. (Citation2002) and similar to the quantitative PCR (qPCR) method of Rogers et al. (Citation2009). High sensitivity was achieved and detection of low spore concentrations was more consistent than the nested PCR method described by Qin et al. (Citation2010), which detected five ascospores per PCR reaction from an ascospore suspension in eight of nine tests, and the equivalent of two to three ascospores per reaction, from inoculated Brassica napus petals in only one in 10 tests. For the field samples tested, our method checked for false negatives, possibly due to very low levels of ascospore DNA in 1 µL, by running the PCR a second time with a larger volume of DNA (3 µL). Further improvements to detection may be made by extracting DNA from both rods from the rotating sampler. In our study, DNA was extracted from only one rod and the other was retained for storage and further testing if required.
Due to the small size of the product amplified by the primers developed in this study (SIG2598F and SIG2767R), they have potential to be adapted for the development of a qPCR test, which may further enhance the understanding of relationships between environmental conditions and ascosporic inoculum, and subsequent disease development. Quantitative PCR has also been used for the detection of S. sclerotiorum in planta (Yin et al. Citation2009) and has been reported using primers targeting the small sub-unit rRNA for the detection and quantification of ascospores using Burkard volumetric samplers (Parker et al. Citation2014).
This study demonstrated potential for this assay to aid in successful management of white mould, which requires protection of flowers from ascospore infection. Our PCR test was able to detect S. sclerotiorum ascospores on Rotorod spore traps in the pre-flowering stage and prior to row closure. Although there was not an association between ascospore detection and the incidence of white mould near harvest in this study, the presence of ascospores near bloom is a general indication of disease risk (Boland & Hall Citation1987; McDonald & Boland Citation2004). PCR detection of ascospores just preceding bloom could enable growers to target fungicide applications only when inoculum is present.
Acknowledgements
This project was facilitated by Horticulture Australia Ltd, in partnership with AUSVEG and funded by the Vegetable Levy with matching funding from the Australian Commonwealth Government (VG07126). Travel of Dr Gent to Australia was partially funded by the University of Tasmania Visiting Scholar Programme. Thanks also to Craig Palmer (Tasmanian Institute of Agriculture) for technical assistance, Garry McNab (Simplot Australia Pty Ltd) and growers for access to fields.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Abd-Elmagid A, Garrido PA, Hunger R, Lyles JL, Mansfield MA, Gugino BK et al. 2013. Discriminatory simplex and multiplex PCR for four species of the genus Sclerotinia. Journal of Microbiological Methods 92: 293–300. 10.1016/j.mimet.2012.12.020
- Ben-Yephet Y, Bitton S 1985. Use of selective medium to study the dispersal of ascospores of Sclerotinia sclerotiorum. Phytoparasitica 13: 33–40. 10.1007/BF02994435
- Boland GJ, Hall R 1987. Epidemiology of white mould on white bean in Ontario. Canadian Journal of Botany 9: 218–224.
- Carisse O, Tremblay DM, Lévesque C, Gindro K, Ward P, Houde A 2009. Development of a TaqMan real-time PCR assay for quantification of airborne conidia of Botrytis squamosa and management of Botrytis leaf blight of onion. Phytopathology 99: 1273–1280. 10.1094/PHYTO-99-11-1273
- Clarkson JP, Phelps K, Whipps JM, Young CS, Smith JA, Watling M 2004. Forecasting Sclerotinia disease on lettuce: towards developing a prediction model for carpogenic germination of sclerotia. Phytopathology 94: 268–279. 10.1094/PHYTO.2004.94.3.268
- Ekins MG, Aitken EAB, Goulter KC 2002. Carpogenic germination of Sclerotinia minor and potential distribution in Australia. Australasian Plant Pathology 31: 259–265. 10.1071/AP02022
- Falacy JS, Grove GG, Mahaffee WR, Galloway H, Glawe DA, Larsen RC et al. 2007. Detection of Erysiphe necator in air samples using the polymerase chain reaction and species-specific primers. Phytopathology 97: 1290–1297. 10.1094/PHYTO-97-10-1290
- Freeman J, Ward E, Calderon C, McCartney A 2002. A polymerase chain reaction (PCR) assay for the detection of inoculum of Sclerotinia sclerotiorum. European Journal of Plant Pathology 108: 877–886. 10.1023/A:1021216720024
- Gent DH, Nelson ME, Farnsworth JL, Grove GG 2009. PCR detection of Pseudoperonospora humuli in air samples from hop yards. Plant Pathology 58: 1081–1091. 10.1111/j.1365-3059.2009.02143.x
- Gugel RK, Morall RAA 1986. Inoculum-disease relationships in Sclerotinia stem rot of rapeseed in Saskatchewan. Canadian Journal of Plant Pathology 8: 89–96. 10.1080/07060668609501848
- Harikrishnan R, del Río LE 2008. A logistic regression model for predicting risk of white mold incidence on dry bean in North Dakota. Plant Disease 92: 42–46. 10.1094/PDIS-92-1-0042
- Hartill WFT 1980. Aerobiology of Sclerotinia sclerotiorum and Botrytis cinerea in New Zealand tobacco crops. New Zealand Journal of Agricultural Research 23: 259–262. 10.1080/00288233.1980.10430796
- Jones SJ, Gent DH, Pethybridge SJ, Hay FS 2011. Spatial characteristics of white mould epidemics and the development of sequential sampling plants in Australian bean fields. Plant Pathology 60: 1169–1182. 10.1111/j.1365-3059.2011.02466.x
- Klosterman SJ, Anchieta A, McRoberts N, Koike ST, Subbarao KV, Voglmayr H et al. 2014. Comparing spore traps and quantitative PCR assays for detection of the downy mildew pathogens of spinach (Peronospora effusa) and beet (Peronospora schachtii). Phytopathology 104: 1349–1359. 10.1094/PHYTO-02-14-0054-R
- McCartney HA, Lacey ME 1991. The relationship between the release of ascospores of Sclerotinia sclerotiorum, infection and disease in sunflower plots in the United Kingdom. Grana 30: 486–492. 10.1080/00173139109432015
- McCartney HA, Lacey ME 1999. Timing and infection of sunflowers by Sclerotinia sclerotiorum and disease development. Aspects of Applied Biology 56: 151–156.
- McDonald MR, Boland GJ 2004. Forecasting diseases caused by Sclerotinia spp. in eastern Canada: fact or fiction? Canadian Journal of Plant Pathology 26: 480–488. 10.1080/07060660409507168
- O’Malley T 2012. Epidemiology and management of pyrethrum flower diseases. PhD dissertation. Hobart, Australia, University of Tasmania.
- Parker ML, McDonald MR, Boland GJ 2014. Evaluation of air sampling and detection methods to quantify airborne ascospores of Sclerotinia sclerotiorum. Plant Disease 98: 32–42. 10.1094/PDIS-02-13-0163-RE
- Qin L, Fu Y, Xie J, Cheng J, Jiang D, Li G et al. 2010. A nested-PCR method for rapid detection of Sclerotinia sclerotiorum on petals of oilseed rape (Brassica napus). Plant Pathology 60: 271–277. 10.1111/j.1365-3059.2010.02372.x
- Rogers SL, Atkins SD, West JS 2009. Detection and quantification of airborne inoculum of Sclerotinia sclerotiorum using quantitative PCR. Plant Pathology 58: 324–331. 10.1111/j.1365-3059.2008.01945.x
- Schwartz HF, Steadman JR 1978. Factors affecting sclerotium populations of, and apothecium production by Sclerotinia sclerotiorum. Phytopathology 68: 383–388. 10.1094/Phyto-68-383
- Steadman JR, O’Keefe D 1999. The blue plate test: a simple method to indicate the potential for white mold in dry beans and soybeans. In: Nelson BD, Gulya TJ eds. Proceedings of the 1998 International Sclerotinia Workshop, Fargo, ND, USA, 9–12 September 1998: 28. British Society of Plant Pathology and International Society of Plant Pathology.
- Sutton DC, Deverall BJ 1983. Studies on infection of bean (Phaseolus vulgaris) and soybean (Glycine max) by ascospores of Sclerotinia sclerotiarum. Plant Pathology 32: 251–261. 10.1111/j.1365-3059.1983.tb02832.x
- Turkington TK, Morall, RAA 1993. Use of petal infestation to forecast Sclerotinia stem rot of canola: the influence of inoculum variation over the flowering period and canopy density. Phytopathology 83: 682–689. 10.1094/Phyto-83-682
- Williams GH, Western JH 1965. The biology of Sclerotinia trifoliorum Erikss. and other species of sclerotium-forming fungi. 1. Apothecium formation from sclerotia. Annals of Applied Biology 56: 253–260. 10.1111/j.1744-7348.1965.tb01233.x
- Yin Y, Ding L, Liu X, Yang J, Ma Z 2009. Detection of Sclerotinia sclerotiorum in planta by a real-time PCR assay. Journal of Phytopathology 157: 465–469. 10.1111/j.1439-0434.2009.01543.x
- Young CS, Fawcett LE, Anthony SG, Smith JA, Watling M, Clarkson JP et al. 2005. Forecasting Sclerotinia disease in field-grown lettuce. Plant Pathology 53: 387–397. 10.1111/j.1365-3059.2004.01018.x