
ABSTRACT
Biogenic hydroxyapatite (bio-HA) has the potential for radionuclide capture and remediation of metal-contaminated environments. Biosynthesis of bio-HA was achieved via the phosphatase activity of a Serratia sp. supplemented with various concentrations of CaCl2 and glycerol 2-phosphate (G2P) provided at pH 7.0 or 8.6. Presence of hydroxyapatite (HA) was confirmed in the samples by X-ray powder diffraction analysis. When provided with limiting (1 mM) G2P and excess (5 mM) Ca2+ at pH 8.6, monohydrocalcite was found. This, and bio-HA with less (1 mM) Ca2+ accumulated Eu(III) to ∼31% and 20% of the biomineral mass, respectively, as compared to 50% of the mineral mass accumulated by commercial HA. Optimally, with bio-HA made at initial pH 7.0 from 2 mM Ca2+ and 5 mM G2P, Eu(III) accumulated to ∼74% of the weight of bio-HA, which was equal to the mass of the HA mineral component of the biomaterial. The implications with respect to potential bio-HA-barrier development in situ or as a remediation strategy are discussed.
Introduction
Anthropogenic radionuclides are responsible for contamination of a range of environments as a result of nuclear activities. Historic examples include leakage from waste storage tanks/ponds (e.g., Hanford, USA or Sellafield sites, UK) as well as arising from large-scale nuclear accidents (e.g., Chernobyl, Ukraine or the Fukushima Daiichi nuclear power plant (FDNPP), Japan) (Johnson Citation2013; Povinec et al. Citation2013; Wallace et al. Citation2012; Yablokov et al. Citation2009;). The FDNPP complex presents a continuing challenge for remediation and for radionuclide disposal, while the long-term storage and disposal of nuclear wastes more generally is an area of intense study with respect to a deep geological disposal facility (GDF).
Intermediate-level waste contains substantial amounts of biodegradable cellulosic material (Bassil et al. Citation2015). However, a deep GDF, while containing potential nutrients, would comprise an extreme environment with stresses to the local biota including, in addition to high radiation, highly alkaline waters arising from contact with cementitious material used as backfilling around steel containers containing stored waste (Berner Citation1992). It has been assumed that the GDF environment is too hostile to support microbial growth, although some microorganisms can tolerate such extreme conditions (Bassil et al. Citation2015; Chicote et al. Citation2004; Rizoulis et al. Citation2012). An alkali degradation product of cellulose, isosaccharinic acid (ISA) in addition to chelating agents like EDTA and nitrilotriacetic acid (used in decontamination processes) presents available nutrients for alkaliphilic bacteria; these compounds, if allowed to persist, would complex with radionuclides and assist their mobility (Bassil et al. Citation2015; Bouchard et al. Citation2006; Glaus and Van Loon Citation1999; Van Loon and Glaus Citation1997). Once resaturated with groundwater, the GDF might encourage the proliferation of alkaliphilic microorganisms that can degrade ISA at high pH and hence potentially retard radionuclide migration as a first line of defense (Bassil et al. Citation2015; Kuippers et al. Citation2015). However, the pH will fall with time and distance from the GDF; hence, the formation of barriers to local, as well as wider nuclide dispersion at less extreme alkaline pH, as well as in neutral environments in other situations like the FDNPP, or in groundwaters, should also be considered.
The immobilization of metals and radionuclides in the presence of phosphate minerals offers a potential strategy for radionuclide sequestration in situ (Wright et al. Citation2011). These materials have advantages as they are generally poorly soluble and, with calcium, they readily occur as apatites, e.g., hydroxyapatite [Ca5(PO4)3(OH)]. Such minerals can be made biogenically by stimulation of activity of microorganisms (Wright et al. Citation2011) and/or by introduction of bacteria that have particular properties. For example, hydroxyapatite (HA), formed via the activity of a naturally occurring soil Serratia sp., removed Sr2+ and Co2+ from aqueous solution (Handley-Sidhu et al. Citation2011a), artificial groundwater (Handley-Sidhu et al. Citation2011a) and also sea water (Handley-Sidhu et al. Citation2016) where, in laboratory tests, chemically manufactured HA had lower effect (Handley-Sidhu et al. Citation2011b).
The HA-mineral biogenesis by this model organism relies upon the enzymatic liberation of inorganic phosphate by the bacterium at the cell surface and within the extracellular polymeric matrix (EPM) via cleavage of a phosphate donor substrate (e.g., glycerol-2-phosphate). This promotes metal precipitation exterior to the bacterial surface within the EPM via cell surface nucleation sites and subsequent biomineral growth at these structured locations (Bonthrone et al. Citation2000; Gangappa et al. Citation2016; Handley-Sidhu et al Citation2011b; Handley-Sidhu et al. Citation2016).
Calcium apatite is a family of compounds with the general chemical structure Ca10-nXn (PO4)6-mYmZ2; X and Y represent cations (e.g., Sr2+, Na+, Pb2+ and Cd2+ etc.) and anions (e.g., HPO42− and CO32− etc.) that can substitute for PO4 3− groups in the apatite structure, while Z can be OH−, F−, Cl−, or Br− (Elliott Citation1994). Biogenic hydroxyapatite (bio-HA) has been shown to incorporate Sr2+ and Co2+ within the bulk inorganic amorphous phase (Handley-Sidhu et al. Citation2011a, b), whereas Eu3+ localizes in amorphous grain boundaries (Handley-Sidhu et al. Citation2014; Holliday et al. Citation2012).
Eu(III) has been used as a convenient “surrogate” for trivalent actinide elements. In wastes, the isotope 241Pu (t½ = 14.3 y) dominates the radioactive inventory, giving the daughter 241Am (t1/2 = 433 y), the most common form of which is Am(III) (Cantrall and Felmy Citation2012). While Am(III) has some enhanced mobility compared to Pu(IV) (Cantrall and Felmy Citation2012), a long-term concern is the occurrence of its daughter 237Np(V) (t1/2 = 2.14 × 106 y) since the NpO2+ ion has a high environmental mobility with behavior similar to that of cations like Na+ (Poinssot and Geckeis Citation2012); hence, immobilization of Am(III) would retard the environmental exposure of Np. A previous study showed similar accumulation of both Eu(III) and the actinide Cm(III) by bio-HA (Holliday et al. Citation2012).
A smaller crystallite size of bio-HA as compared to its commercial counterpart was suggested to favor enhanced sequestration of metal ions that bind at grain boundaries as compared to those that accumulate solely within the bulk amorphous phase, attributed to a higher proportion of surface sites in smaller crystals (Gangappa et al. Citation2016; Handley-Sidhu et al. Citation2014). The aims of this study were to evaluate the scope for using bio-HA synthesized by the model organism Serratia sp. as a material for sequestration of Eu(III) when synthesized under various conditions prior to Eu(III) exposure and to evaluate whether factors other than HA crystallite size may be involved in metal sequestration by bio-HA. The results are discussed with a longer term view to conditions relevant to in situ barriers or for ex situ water filtration applications.
Materials and methods
Preparation of biomass and biominerals
Serratia NCIMB 40259, used by permission of Isis Innovation, Oxford, was cultured under carbon (lactose)-limited continuous condition at 28−30°C in an airlift fermenter; the culture was monitored by measurement of phosphatase activity and cell density of free cells in the vessel outflow as described previously (Gangappa et al. Citation2016; Ledo et al. Citation2008; Paterson-Beedle and Macaskie Citation2005). The outflow cells were collected by centrifugation, washed twice in distilled water and stored as pellets at 4°C until use.
Biominerals were synthesized in flasks containing 300 ml of TAPSO/NaOH buffer (50 mM, pH 8.6 or 7.0; pKa at 25°C is 7.60) and cells of Serratia sp. (resuspended to OD600 = 1.0). Flasks were dosed with concentrated solutions of trisodium citrate, CaCl2, and glycerol-2-phosphate (G2P) as shown in (8 daily doses) and shaken (100 rpm) at 28−30°C throughout. The biomaterials were harvested by centrifugation, washed twice in distilled H2O, dried to constant weight (50−60°C), and manually ground using a pestle and mortar. The ground samples were immediately sieved (Test Sieve, PAT No-667924, mesh no. 40, aperture 420 µm). Hydroxyapatite (HA) type −1 (Sigma, EC No 215-145-7) was used for comparative studies. Each 300-ml preparation contained 150 mg of dry biomass; with reference to a previously determined OD600 to dry weight conversion, the HA mineral content could be calculated by subtracting the known dry weight of biomass from the dry weight of the total biomineral sample.
Table 1. HA samples examined in this study.
Characterization of biominerals by X-ray powder diffraction
The dry biomineral powder samples and commercial HA were analyzed using X-ray powder diffraction (XRD; Bruker D8 Advanced X-ray diffractometer; 20° to 70° 2θ; 0.020° step size; 104 sec step time; Cu Kα radiation). The crystallite size was estimated from obtained peaks (2θ) using the Scherrer equation, with Bruker-AXS EVA software being used to identify the biomineral phases.
Accumulation of Eu(III) by biominerals
Solutions of Eu (NO3)3 were prepared in distilled water (≤15.0 MΩ/cm) to 0.5 mM (pH 5–6). All tests were carried out using bio-HA made from two independent cell preparations (at different times) and triplicate suspensions for each. Eu(III) uptake by biominerals was tested by suspending 11 mg of each biomineral in 100 ml of 0.5 mM Eu(NO3)3 at room temperature. At intervals (0.5–48 h), samples were harvested by centrifugation. Residual Eu(III) in the supernatant (30 μl sample) was measured by adding 1.97 ml of 1M ammonium acetate buffer (pH 3.3); Eu(III) was visualized by adding (100 µl) 0.15% w/v (aq.) arsenazo (III) and estimated at A652 with reference to a standard curve, similarly prepared. Under these conditions, Ca(II) is not visualized using arsenazo III as confirmed by preliminary checks. Selected samples were also analyzed by Inductively Coupled Plasma-Optical Emission Spectroscopy (ICP-OES; Agilent 7500ce; University of Plymouth, UK) to determine any release of Ca2+ accompanying Eu3+ accumulation, indicative of ion exchange; no Ca2+ was observed at a limit of detection of 4 µg/L (Gangappa et al. Citation2016). The inorganic phosphate concentration in the solution was measured using the molybdenum blue assay (Handley-Sidhu et al. Citation2011a).
Results and discussion
Synthesis and examination of biominerals
Biominerals were synthesized using Serratia cell suspensions with varied concentrations of Ca2+ and glycerol-2- phosphate (G2P) (). Biomaterials (A–C, made at pH 7.0 and D & E, made at pH 8.6: ) were confirmed as bio-hydroxyapatite (); a full mass balance was not attempted. Although 40-mM G2P was provided to samples A and D over the period of 8 days (), negligible residual inorganic phosphate (Pi) was found in the solutions. Hence, samples A and D (total of 40 mM G2P) could contain potentially 5-fold more phosphate-mineral () than the remaining samples, which were dosed with only 1-mM G2P daily (). This was confirmed. In addition, both these samples were supplied with a total of 16 mM Ca2+, and hence, the calculated mineral content was higher (by ∼ 30%) than that accountable as HA alone (ratio of Ca:P in HA is 1:1.67); the higher mass generation was confirmed for sample A only (). This may be explained by accumulation of polyphosphate (polyP) by the cells. A previous study (Lugg Citation2005) noted that the cells contained polyP deposits [as well as accumulations of poly-hydroxybutyrate (PHB) (Lugg et al. Citation2008); no nitrogen source was provided, and hence, the cells could not grow], and they thus conserved carbon and energy by accumulation of the storage materials PHB and Na-polyP, respectively. Storage of polymeric reserves is well known in microorganisms; a more detailed study will be reported in a subsequent publication, but electron opaque cellular inclusion bodies were clearly visible within Serratia cells in addition to PHB globules (Lugg Citation2005; Lugg et al. Citation2008) and in addition to the extracellular mineral deposits (see supplementary information, Figure S1). Evidence was also visible for this in previous work using cells from Serratia biofilm (Ledo et al. Citation2008), but its significance was not noted or discussed.
Figure 1. X-ray powder diffraction analysis of bio-hydroxyapatite (bio-HA) samples. (a). X-ray powder patterns for bio-HA samples A, B, C, D, and E as shown in . Bio-HA samples were identified by their matching peak positions with commercial HA as indicated by arrows. (b) X-ray powder pattern of sample F, showing peaks of monohydrocalcite (asterisked). Peaks for monohydrocalcite were identified using the selected powder diffraction data using Bruker-AXS EVA software as described in Materials and Methods. Gray dotted lines indicate peaks matching commercial HA, thus suggesting a mixed biomineral (HA/monohydrocalcite). Peaks for the latter (asterisked) were seen only in sample made at pH 8.6 ().
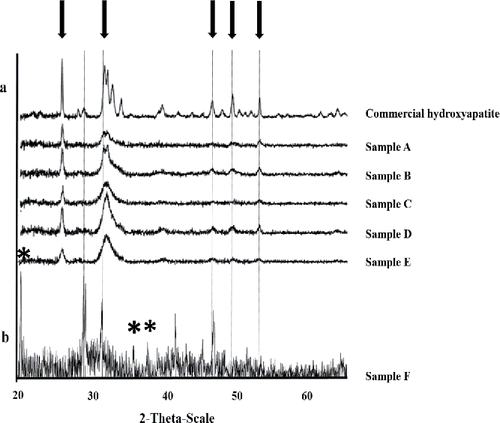
Table 2. Materials from samples made at pH 7 with daily provision of 5 mM and 1 mM G2P.
Hydroxyapatite is most stable at pH > 4, and it normally precipitates at pH above 8 (Boudeville et al. Citation1999). However, another study (Yong et al. Citation2004) showed that once nucleated onto bacterial cells, bio-HA deposition proceeded at pH 7, i.e., the initial nucleation step, influenced by local micro-environmental factors is the limiting factor at neutral pH, and not the crystal growth. Analysis by 31P NMR showed the participation of phosphate groups within the EPM in the nucleation of metal phosphate onto the cells (Bonthrone et al. Citation2000). Therefore, the bio-HA made at pH 7.0 and 8.6 was compared in the current study.
XRD patterns are shown for bio-HA samples A–E () and commercial HA, where the close matches indicate the presence of a hydroxyapatite material, with the ill-defined peaks of bio-HA being attributed to nanocrystalline or amorphous components. The nominal crystallite size for each bio-HA sample (mean 21 nm; ) was less than half the size of commercial HA, with no difference in crystallite size between samples made from solution at pH 7.0 or 8.6 (). Note, however, that the solution pH is not the local pH at the site of mineral formation as this occurs within a reaction “compartment” bounded by the extracellular polymeric material surrounding the cells (see supplementary information, Figure S1). This hydrated polymer contains chemical groups (e.g., carboxyl, phosphate) that would provide a local buffering function (see discussion below).
(sample F) shows the XRD pattern for the biomineral synthesized with a daily dose of 5 mM Ca2+, 1 mM G2P, and 2 mM citrate at an initial pH of 8.6 (). The powder pattern shows the material to contain monohydrocalcite (CaCO3. H2O; , asterisked), but some peaks can be attributed to hydroxyapatite (HA), suggesting the presence of a mixed HA/calcite material (). The mass of dry mineral made at pH 8.6 with a total of 8 mM G2P and 40 mM Ca2+ (sample F: 608 mg) was higher than its counterpart made from solution at pH 7.0 (sample C: 424 mg) (i.e., total mass of each harvest minus the biomass dry weight). The synthesis of higher mass of biomaterial in ‘sample F’ can be attributed to the formation of additional mineral as calcite, this reaction being possibly accelerated via production of CO2 via metabolism of citrate (via the tricarboxylic acid [TCA] cycle), and the formation of soluble bicarbonate ion and then carbonate as CO2 moves outward to the alkaline bulk solution along the pH gradient surrounding the cell within the layer of extracellular polymeric material (Figure S1). In contrast to sample F, no crystalline calcium carbonate was evident in the counterpart mineral made in solution at pH 7.0 (sample C; ), but this does not preclude the formation of amorphous CaCO3 in the latter.
Microbial precipitation of calcium carbonate (calcite, CaCO3) involves four main factors: Ca2+ concentration, dissolved inorganic carbon, alkaline pH, and availability of nucleation sites (Lauchnor et al. Citation2013). Continued cellular metabolism (and hence biogenic CO2 production) at pH 8.6 under these conditions was previously indicated by the ability of the Serratia cells to accumulate polyhydroxybutyrate (Lugg Citation2005; Lugg et al. Citation2008). In addition, the presence of citrate has been reported to favor the calcite crystallization (Tobler et al. Citation2015). The citrate would complex with Ca2+ and thereby reduce the availability of free calcium ions in the medium for rapid precipitation with phosphate in the bulk solution. On the other hand, Serratia sp. utilizes citrate readily; the cells had been preconditioned by growth under carbon limitation and would sequester citrate (as the calcium chelate) and metabolize that into a PHB “sink,” thereby lowering the local concentration of citrate in the immediate space around the periphery of the cell (and also releasing free Ca2+ intracellularly as citrate is metabolized). This would, in turn, promote the movement of more calcium citrate complex down the concentration gradient toward the cell surface, thereby accumulating Ca2+ for potential calcite precipitation. Yates and Robbins (Citation1999) provided evidence for this mechanism using radioactive labeled Ca2+.
According to Hammes and Verstraete (Citation2002), high extracellular calcium and low extracellular proton concentration (i.e. high pH) is a typical condition for microbial CaCO3 precipitation. An alkaline pH would present a stressful environment for bacteria; as a result of a complementary Ca2+/2H+ electrochemical gradient across the membrane, a passive influx of calcium would lead to additional intracellular Ca2+ build-up (over and above that released intracellularly from the citrate complex), and proton expulsion may occur to counterbalance this (Hammes and Verstraete Citation2002), creating a localized pH downshift externally and increasing the pH of the cytoplasm. This condition would be deleterious to the bacterial cells as a high internal Ca2+ would disrupt intracellular calcium-regulated signal processes and promote a higher intracellular pH (via the reduction of the proton pool). In response to such conditions, bacteria may then switch to active discharge of intracellular Ca2+ and import excess extracellular protons, promoting a localized increase in extracellular pH with a simultaneous rise in Ca2+ in the same region, thus presenting a local microenvironment for metal precipitation (Hammes and Verstraete Citation2002). The dissolved inorganic carbon (“metabolic” CO2) would form soluble (bi) carbonate ions; at alkaline pH, carbonate ions tend to react with Ca2+, leading to calcium carbonate (CaCO3) crystals (Lauchnor et al. Citation2013). A combination of these factors would result in dynamic pH and ion gradients within the “housing” matrix of the EPM. The result at pH 8.6 is illustrated in supplementary information (Figure S1) where the HA biomineral is evident as dense needle-like HA deposits close to the cell body with more amorphous material visible in the outermost layers of the EPM. A progression of amorphous to crystalline material is suggested where isolated HA crystals can be seen surrounded by amorphous material (Figure S1).
Eu(III) accumulation by biominerals
Bio-hydroxyapatite made at initial pH 7 from 2 mM Ca2+ and 5 mM G2P was the most effective for Eu(III) sequestration (), comprising ∼74% of the biomaterial mass (sample A in ) corresponding to ∼1 mg Eu(III) per mg of HA mineral. In contrast, bio-HA made with 1 mM of each Ca2+ and G2P (sample B) showed ∼55% of Eu(III) sequestration () i.e., comparable to commercial HA. In contrast, the bio-HA synthesized from 5 mM Ca2+ and 1 mM G2P (sample C) showed ∼23% sequestration of Eu(III) () or 0.3 mg Eu(III) per mg of HA, which was identical to that obtained in the counterpart sample (sample F) made at pH 8.6. Formation of crystalline calcite was not apparent from any samples made in solution at pH 7 or those at pH 8.6 with 1 mM or 2 mM Ca2+ (; ), but amorphous CaCO3 cannot be excluded since XRD detects only crystalline material.
Figure 2. Eu(III) uptake (% of the biomineral) by biomineral samples A–F made as described in Materials and Methods with daily additions as shown in from solutions at initial (a) pH 7 and (b) 8.6. Europium uptake by commercial hydroxyapatite (HA) is shown for comparison (gray bar). Errors (SEM) were generally within 5% of the mean throughout.
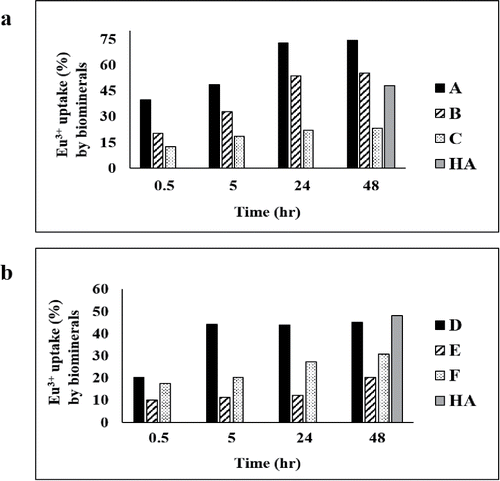
The Eu(III) accumulation capacity of bio-HA prepared from solution at pH 8.6 under excess G2P (sample D in ) was ∼1.7 times lower than that of bio-HA prepared at pH 7 (sample A) with similar substrate concentrations, although the crystallite size () of all samples was comparable. This indicates that factors other than solely crystallite size influence the accumulation capacity.
As reported by Handley-Sidhu et al. (Citation2014), accumulation of Eu(III) was found at grain boundaries (where the size of the crystallites would be influential), while substitution of Ca with neodymium and dysprosium was also observed in earlier work, with the Nd and Dy ions occupying mainly Ca(2) positions of HA (Getman et al. Citation2005) at a tricalcium phosphate layer of the crystallite surface (Handley-Sidhu et al. Citation2014). The nature of the bio-HA material was discussed previously (Ledo et al. Citation2008).
Sample E (synthesized using 1 mM each of calcium and G2P at initial pH 8.6) showed ∼3 fold less europium accumulation than its counterpart (sample B) made at initial pH 7.0 (), hence, the effect of pH of sample manufacture was apparent, whereas samples made with 5 mM Ca2+ accumulated Eu(III) identically but poorly, irrespective of the manufacturing pH (Sample C and F in or ). On the other hand, bio-HA was shown previously to be slightly calcium-deficient (Ca/P ratio of 1:1.61) (Ledo et al. Citation2008), and with a defective HA crystal lattice as suggested by FTIR (Ledo et al. Citation2008) while also containing Na+ and with the structure modified by the presence of organic impurities from the EPM (Ledo et al. Citation2008). Regardless of the pH of synthesis, material made using excess Ca2+ gave only 0.3 mg Eu(III) accumulation per mg of HA mineral; it may be suggested, therefore, that a Ca deficiency in the biomineral may assist in formation of the Eu-substituted form; however, since these accumulations were lower than those for commercial HA, a further study was not made.
Europium is present as Eu (III) below pH 7.0 whereas Eu (OH)2+ is predominant between pH 8.0 and 9.0 (Sastri et al. Citation2003). Ledo et al. (Citation2008) noted that the presence of organic contaminants from the EPM may influence the structure of the bio-HA. A pH of 7 is closer to the pKa of carboxyl groups and hence, assuming a pH gradient from the solution toward the cell surface, a larger proportion of the extracellular polymer would be in the protonated form nearer to the cell surface at pH 7 than at pH 8.6 during the HA formation process; this may influence the morphology of the HA forming at pH 7, as compared to material “housed” within more dissociated polymer at alkaline pH. Material made under a more alkaline pH gradient (exterior alkaline) would have a larger proportion of dissociated groups toward the outside of the EPM, with Na+ (from the G2P and citrate salts used) used as the counterion, which may account for the source of Na+ suggested as a component of the bio-HA (Ledo et al. Citation2008) and possibly tending to have a cation-competing effect against incoming Eu(III) for incorporation of the latter into HA.
Due to the alkaline pH used during the HA synthesis in samples D-F, the associated fraction of the EPM carboxyl groups would be in the form of –COONa; on transfer into aqueous Eu(III) solution, Na+ release may result in local alkalinization; hence, the speciation of Eu3+ would shift locally toward Eu(OH)2+. In contrast, HA material that was made at pH 7 would tend to yield protons from its carboxylate surroundings in water rather than Na+, and the mixture would tend to retain Eu(III) as Eu3+. It is possible that Eu3+ interacts differently with HA as compared to Eu(OH)+ or Eu(OH)2. A more detailed structural comparison of the bio-HA made at the two pH values is warranted, and confirmation of the formation of hydrolysis products of Eu(III) via local alkalinization is attributable to Na+ release from EPM (rather than protons). It is important to note that XRD provides no information about the amorphous component of HA, and it is not quantitative. Even without the added dynamic microbial interactions with Ca2+, citrate, and G2P (above) and given that this is a three-phase system (hydrated EPM, solid mineral and bulk water) with various spatial gradients (e.g., see example of Figure S1), the chemical processes occurring within the EPM hydrogel would be difficult to delineate. MRI, which has the capability to map pH gradients in situ, has a spatial resolution of only ∼ 100 microns (Macaskie et al. Citation2005), whereas the bacterial cell together with its EPM is ∼2–3 µm when dehydrated (Figure S1) and ∼5–10 µm when fully hydrated in a biofilm (Macaskie et al. Citation2005).
The bio-HA crystallite size was comparable throughout and was ∼half that of the commercial HA (). Handley-Sidhu et al. (Citation2014) suggested that the smaller crystallite size of bio-HA may have an advantage for high Eu(III) sorption as the latter substitutes at the Ca(2) and/or the Ca(3) position of tricalcium phosphate, a known component of HA grain boundaries; the higher proportion of surface sites in smaller crystals may be advantageous, but this does not explain the different observations using HA made at the two pH values. At pH 7, samples A and B (made from 2 and 1 mM Ca2+) accumulated Eu(III) in a time-dependent way to reach capacity after 24 h (). In contrast, sample D (made at an initial pH of 8.6) reached capacity after only 5 h with no further increase in the sequestration of Eu(III) (). At this 5-h stage, the accumulation by samples A and D was comparable (∼50%), i.e., there was no evidence that pH used for HA synthesis had an effect; hence, the subsequent increase seen in sample A (which is responsible for its superiority as compared to commercial HA: ) may be attributed to a second mechanism, which was lacking in the sample made at pH 8.6, but this was not tested further. This secondary phase in sample A (24–48 h; ) was associated with the enhanced accumulation seen over and above that of the commercial HA and the other bio-HA samples. These results suggest that Eu(III) sequestration is dependent on factors other than simply the crystallite size since this was comparable throughout ().
Biomineral produced from solution at pH 8.6 under excess Ca2+ (sample F; ) showed Eu(III) accumulation to ∼31% after 48 h (), corresponding to only 0.3 mg Eu(III) per mg of mineral. This biomineral was identified as a possible monohydrocalcite although some HA was still apparent (). However, Lakshtanove and Stipp (Citation2004) reported that Eu3+ has a strong affinity toward calcite, and its sorption does not rely on precipitation rate and particle size. In contrast, the present study suggests that the formation of a putative calcite/HA biomineral is disadvantageous to the overall uptake of Eu(III) (see above; the presence of amorphous calcite at pH 7 cannot be ruled out), and while calcite mineral has been reported to have good potential for trapping problematic trace metals and radionuclides (Handley-Sidhu et al. Citation2013), genesis of calcite alongside HA is possibly not beneficial toward Eu(III) uptake.
Conclusion
The bio-HA (sample A) prepared at initial pH 7.0 using Serratia cells with 2 mM Ca2+ and 5 mM G2P showed ∼74% Eu3+ (mass/mass biomaterial) uptake, while bio-HA (sample D) synthesized similarly at pH 8.6 showed ∼45% Eu3+ accumulation, suggesting the potential of former bio-HA material for use in radionuclide clean up technologies. This may also be valuable in the recovery of rare earth elements from mine and wastewaters.
Monohydrocalcite was also formed when Serratia cells were challenged with excessive Ca2+ at alkaline conditions (pH 8.6). This material showed ∼31% (mass/mass biomaterial) of Eu3+ uptake under conditions where commercial HA accumulated Eu (III) to 50% of its mass, and bio-HA 74%. The high uptake of bio-HA made under excess G2P and initial pH 7.0 was attributed to factors other than crystallite size.
Figure S1.docx
Download MS Word (1.8 MB)Acknowledgments
The authors acknowledge, with thanks, the assistance of Ms S. Singh and Dr A.J. Murray for the provision of planktonic bacterial cells used in one preparation in this study. The electron micrograph shown in supplementary information was obtained by Ms. A. Dennis as part of an undergraduate project.
Funding
This project was supported by the BBSRC (Grant No. 6/E11940), NERC (Grant Nos NE/L014076/1 and NE/L012537/1) and EPSRC (Grant nos EP/C548809/1 and EP/M012719/1). Serratia sp. NCIMB 40259 was made available for this study by kind permission of Isis Innovation, Oxford, UK.
Related Research Data
References
- Bassil NM, Bryan N, Lloyd JR. 2015. Microbial degradation of isosaccharinic acid at high pH. The ISME journal 9:310–320.
- Berner UR. 1992. Evolution of pore water chemistry during degradation of cement in a radioactive waste repository environment. Waste Manag 2:201–219.
- Bonthrone KM, Quarmby J, Hewitt CJ, Allan VJM, Paterson-Beedle M, Kennedy JF, Macaskie LE. 2000. The effect of the growth medium on the composition and metal binding behaviour of the extracellular polymeric material of a metal-accumulating Citrobacter sp. Environ Technol 21:123–134.
- Bouchard J, Methot M, Jordan B. 2006. The effects of ionizing radiation on the cellulose of woodfree paper. Cellulose 13:601–610.
- Boudeville P, Serraj S, Leloup JM, Margerit J, Pauvert B, Terol A. 1999. Physical properties and self-setting mechanism of calcium phosphate cements from calcium bis-dihydrogenophosphate monohydrate and calcium oxide. J Mater Sci: Mater Med 10:99–109.
- Cantrall KJ, Felmy AR. 2012. Plutonium and Americium Geochemistry at Hanford: A Site-Wide Review. Richland Washington: Pacific Northwest National Laboratory.
- Chicote E, Moreno D, Garcia A, Sarro I, Lorenzo P, Montero F. 2004. Biofouling on the walls of a spent nuclear fuel pool with radioactive ultrapure water. Biofouling 20:35–42.
- Elliott JC. 1994. Structure and chemistry of the apatites and other calcium orthophosphates. Elsevier, Amsterdam.
- Gangappa R, Yong P, Singh S, Mikheenko I, Murray AJ, Macaskie LE. 2016. Hydroxyapatite biosynthesis by a Serratia sp. and application of nanoscale bio-HA in the recovery of strontium and europium. Geomicrobiol J 33:267–273.
- Getman EI, Loboda SN, Tkachenko TV, Ignatov AV, Zabirko TF. 2005. Substitution of calcium with neodymium and dysprosium in hydroxyapatite structure. Funct Mat 12:6–10.
- Glaus MA, Van Loon LR, Achatz S, Chodura A, Fischer K. 1999. Degradation of cellulosic materials under the alkaline conditions of a cementitious repository for low and intermediate level radioactive waste: Part I: Identification of degradation products. Anal Chim Acta 398:111–122.
- Hammes F, Verstraete W. 2002. Key roles of pH and calcium metabolism in microbial carbonate precipitation. Rev Environ Sci Biotechnol 1:3–7.
- Handley-Sidhu S, Hriljac JA, Cuthbert MO, Renshaw JC, Pattrick RAD, Charnock JM, Stolpe B, Lead JR, Baker S, Macaskie LE. 2014. Bacterially produced calcium phosphate nanobiominerals: sorption capacity, site preferences, and stability of captured radionuclides. Environ Sci Technol 48:6891–6898.
- Handley-Sidhu S, Mullan TK, Grail Q, Albadarneh M, Ohnuki T, Macaskie LE. 2016. Influence of pH, competing ions, and salinity on the sorption of strontium and cobalt onto biogenic hydroxyapatite. Scientific Reports 6.
- Handley-Sidhu S, Renshaw JC, Moriyama S, Stolpe B, Mennan C, Bagheriasl S, Yong P, Stamboulis A, Paterson-Beedle M, Sasaki K, Pattrick RAD, Lead JR, Macaskie LE. 2011a. Uptake of Sr2+ and Co2+ into biogenic hydroxyapatite: implications for biomineral ion exchange synthesis. Environ Sci Technol 45:6985–6990.
- Handley-Sidhu S, Renshaw JC, Yong P, Kerley R, Macaskie LE. 2011b. Nano-crystalline hydroxyapatite bio-mineral for the treatment of strontium from aqueous solutions. Biotechnol Lett 33:79–87.
- Handley-Sidhu S, Sham E, Cuthbert MO, Nougarol S, Mantle M, Johns ML, Macaskie LE, Renshaw JC. 2013. Kinetics of urease mediated calcite precipitation and permeability reduction of porous media evidenced by magnetic resonance imaging. Int J Environ Sci Technol 10:881–890.
- Holliday K, Handley-Sidhu S, Dardenne K, Renshaw J, Macaskie LE, Walther C, Stumpf TA. 2012. New incorporation mechanism for trivalent actinides into bioapatite: A TRLFS and EXAFS study. Langmuir 28:3845–3851.
- Johnson E. Reuters. Accessed Feb 23, 2013. Radioactive waste leaking from six tanks at Washington state nuclear site.www.reuters.com/article/2013/02/24/us-usa-nuclear-leak-idUSBRE91L19G20130224.
- Kuippers G, Bassil NM, Boothman C, Bryan N, Lloyd JR. 2015. Microbial degradation of isosaccharinic acid under conditions representative for the far field of radioactive waste disposal facilities. Min Mag 79:1443–1454.
- Lakshtanov LZ, Stipp SLS. 2004. Experimental study of europium (III) coprecipitation with calcite. Geochim Cosmochim Acta 68:819–827.
- Lauchnor EG, Schultz LN, Bugni S, Mitchell AC, Cunningham AB, Gerlach R. 2013. Bacterially induced calcium carbonate precipitation and strontium coprecipitation in a porous media flow system. Environ Sci Technol 47:1557–1564.
- Ledo HM, Thackray AC, Jones IP, Marquis PM, Macaskie LE, Sammons RL. 2008. Microstructure and composition of biosynthetically synthesized hydroxyapatite. J Mater Sci: Mater Med 19:3419–3427.
- Lugg H. 2005. A study of extracellular calcium phosphate biomineralisation and intracellular inclusion body formation by Serratia sp. N14. PhD thesis, University of Birmingham, UK.
- Lugg H, Sammons RL, Marquis PM, Hewitt CJ, Yong P, Paterson-Beedle M, Redwood MD, Stamboulis A, Kashani M, Jenkins M, Macaskie LE. 2008. Polyhydroxybutyrate accumulation by a Serratia sp. Biotechnol Lett 30:481–491.
- Macaskie LE, Yong P, Paterson-Beedle M, Thackray AC, Marquis PM, Sammons RL, Nott KP, Hall LD. 2005. A novel non line-of-sight method for coating hydroxyapatite onto the surfaces of support materials by biomineralization. J Biotechnol 118:187–200.
- Paterson-Beedle M, Macaskie LE. 2005. Use of PhoN phosphatase to remediate heavy metals. Microb Processes Prod, Humana Press, New York, p413–436.
- Poinssot C, Geckeis H. 2012. Eds. Radionuclide Behaviour in the Natural Environment: Science, Implications and Lessons for the Nuclear Industry. Elsevier.
- Povinec PP, Hirose K, Aoyama M. 2013. Fukushima Accident: Radioactivity Impact on the Environment. Newnes, Elsevier, Amsterdam.
- Rizoulis A, Steele HM, Morris K, Lloyd JR. 2012. The potential impact of anaerobic microbial metabolism during the geological disposal of intermediate-level waste. Min Mag 76:3261–3270.
- Sastri VS, Bunzli JC, Rao VR, Rayudu GVS, Perumareddi JR. 2003. Modern Aspects of Rare Earths and their Complexes. Elsevier, Amsterdam, p878–879.
- Tobler DJ, Rodriguez-Blanco JD, Dideriksen K, Bovet N, Sand KK, Stipp SL. 2015. Citrate effects on amorphous calcium carbonate (ACC) structure, stability, and crystallization. Adv Funct Mats 25:3081–3090.
- Van Loon LR, Glaus MA. 1997. Review of the kinetics of alkaline degradation of cellulose in view of its relevance for safety assessment of radioactive waste repositories. J Environ Polym Degr 5:97–109.
- Wallace SH, Shaw S, Morris K, Small JS, Fuller AJ, Burke IT. 2012. Effect on ground water pH and ionic strength on strontium sorption in aquifer sediments: Implications for 90Sr mobility at contaminated nuclear sites. Appl Geochem 27:1482–1491.
- Wright KE, Hartmann T, Fujita Y. 2011. Inducing mineral precipitation in groundwater by addition of phosphate. Geochem Trans 12:8.
- Yablokov AV, Nesterenko VB, Nesterenko AV. 2009. Chapter III. Consequences of the chernobyl catastrophe for the environment. Ann N.Y. Acad Sci 1181:221–286.
- Yates KK, Robbins LL. 1999. Radioisotope tracer studies of inorganic carbon and Ca in microbially derived CaCO3. Geochim Cosmochim Acta 63:129–136.
- Yong P, Macaskie LE, Sammons RL, Marquis PM. 2004. Synthesis of nanophase hydrox`yapatite by a Serratia sp. from waste-water containing inorganic phosphate. Biotechnol Lett 26:1723–1730.