
Abstract
Pathogenic, biallelic variants in SORD were identified in 2020 as a novel cause for autosomal-recessive Charcot-Marie-Tooth disease (CMT) type 2, an inherited neuropathy. SORD codes for the enzyme sorbitol dehydrogenase. Loss of this enzyme‘s activity leads to an increase of sorbitol in serum. We retrospectively screened 166 patients with axonal neuropathy (predominantly CMT type 2, but including intermediate form of CMT and distal hereditary motor neuropathy (dHMN)) without identified genetic etiology for SORD mutations at a single large German neuromuscular center. Clinical and electrophysiology exam findings were analyzed for genotype–phenotype correlation. Five patients of the total cohort of 166 patients harbored pathogenic variants in SORD (3%). The homozygous frameshift variant c.757delG (p.Ala253Glnfs*27) was the most common (4/5). One additional case carried this variant on one allele only and an additional pathogenic missense variant c.458C > A (p.Ala153Asp) on the other allele. Age of onset ranged from early infancy to mid-twenties, and phenotypes comprised axonal CMT (4) and dHMN (1). Our findings strengthen the importance of screening for pathogenic variants in SORD, especially in patients with genetically unconfirmed axonal neuropathy, especially CMT type 2 and dHMN.
Introduction
Charcot-Marie-Tooth disease (CMT) is a clinically and genetically heterogeneous group of inherited neuropathies that share common characteristics of distal muscle wasting, sensory loss, and decreased or absent deep tendon reflexes (Rossor et al., Citation2013). Broadly, CMT can be classified as demyelinating (CMT1) and axonal (CMT2) according to motor nerve conduction velocity (MCV) and compound motor action potential (CMAP) amplitude in the upper limb nerve.
Recently, biallelic pathogenic variants in the sorbitol dehydrogenase (SORD) gene were identified as a novel and relatively common genetic cause for autosomal-recessive axonal CMT, for intermediate CMT and distal hereditary motor neuropathy (dHMN) (Cortese et al., Citation2020), and denominated as sorbitol dehydrogenase deficiency with peripheral neuropathy (SORDD, OMIM # 618912). SORDD is a slowly progressive disorder affecting peripheral nerves. Clinically, it is characterized by distal muscle weakness mainly affecting the lower limbs and resulting in difficulty walking with significant reduction of walking distance and frequent falls. Distally pronounced sensory impairment in classical axonal and intermediate CMT2 contributes to sensory ataxia and gait instability. Onset of symptoms is usually in the first or second decade of life, although later adult onset has been reported. Nerve conduction velocities and compound action potential amplitudes are consistent with an axonal phenotype, mainly with mixed axonal degeneration and demyelination or with distal motor neuropathy reported less frequently. Other more variable features may include upper limb tremor and scoliosis.
The SORD gene codes for the enzyme sorbitol dehydrogenase (Cortese et al., Citation2020; Jedziniak et al., Citation1981). Most of the pathogenic variants in SORD reported so far are frameshift or splicing variants, indicating a loss of function of sorbitol dehydrogenase. In the polyol pathway, aldose reductase catalyzes the metabolism of glucose to sorbitol and sorbitol dehydrogenase catalyzes the metabolism of sorbitol to fructose (Gabbay, Citation1973; Oates, Citation2002). Mutations in the SORD gene subsequently lead to decreased levels of sorbitol dehydrogenase, which consequently increases sorbitol in the serum. Sorbitol cannot cross cell membranes, and accumulation produces stress on cells by drawing water into insulin-independent tissues (Jedziniak et al., Citation1981).
Numerous findings have implicated increased aldose reductase activity in the pathogenesis of diabetic neuropathy, retinopathy, nephropathy, and macrovascular disease already (Kato et al., Citation2003; Oates & Mylari, Citation1999; Obrosova et al., Citation2002). However, the pathological mechanisms by which genetic changes in SORD cause motor predominant peripheral neuropathy are not yet fully understood. Very recently, Rebelo et al. (Citation2024) published a Sord-knockout rat model (Sord-/-) that provides novel aspects of SORD deficiency. Sord-/- rats mirror the human phenotype and can be used to improve the understanding of pathophysiology and therapeutic options in SORD-associated neuropathy. Sord-/- rats showed a delay in motor performance at 7 months of age, and a predominantly motor neuropathy was characterized with a battery of behavioral tests supplemented by biochemical, physiological, and comprehensive histological examinations. Increased levels of sorbitol were measured in serum, cerebrospinal fluid (CSF), and peripheral nerve of Sord-/- rats, leading to the assumption that toxic accumulation of sorbitol contributes to gradual damage of peripheral nerves and the emergence of the observed phenotype over time. The findings also suggest that osmolarity imbalance leads to further peripheral nerve damage. Moreover, Sord-/- rats displayed increased levels of neurofilament light chain, NfL, a biomarker of axonal degeneration. These observations in the animal model are valuable for preclinical studies and their translation to human studies.
Importantly, experimental treatment with aldose reductase inhibitors normalized intracellular sorbitol levels in patient-derived fibroblasts and in Drosophila melanogaster, and ameliorated the identified motor and eye phenotype in flies in the original publication (Cortese et al., Citation2020).
Our study provides further information on the frequency, genotype, and phenotype of patients with pathogenic variants in SORD screened in a large Northern German neuromuscular cohort. The frequency of SORD variants was calculated to 3% (5/166) with phenotypes including previously unclarified CMT2 (4) and dHMN (1) patients.
Materials and methods
For this study, we retrospectively screened 166 patients with predominantly axonal neuropathy without identified genetic etiology for possible pathogenic SORD variants by next-generation sequencing (NGS) and subsequent Sanger sequencing to specifically amplify SORD and not the pseudogene SORD2P. Clinical and electrophysiology exam findings are reported for genotype–phenotype correlation and further characterization of the disease.
Clinical details
We retrospectively selected all patients with clinical criteria of an underlying axonal neuropathy which included predominantly CMT type 2 and to a lesser extent intermediate and dHMN forms without identified genetic etiology and subsequently screened for pathogenic variants in SORD after discovery of the gene in 2020. Detailed inclusion criteria were: predominantly axonal degeneration, early onset, typical foot deformities or other indicators of longstanding, inherited disease; no underlying etiology in standard neuropathy work-up including extensive serology and lumbar puncture. CMT1 patients were excluded. Selected patients (166 in total) present regularly to our neuromuscular outpatient clinics where clinical examination, electrophysiology, and differential-diagnostic work-up including molecular genetics analyses were performed in-house.
Molecular genetic studies
WES
For sequencing, genomic DNA was extracted from peripheral blood and further captured using the customized neuromuscular disease (NMD) gene panel (Twist Bioscience, San Francisco, CA, USA) according to the manufacturer’s protocol. Paired-end (2 × 150) sequencing was performed on a MiSeq sequencer (Illumina) with a mean target coverage of >30×. Bioinformatic analysis, including alignment to the GRCh37/hg19 human reference genome, variant and copy number variant calling and variant filtering, was performed using Varvis software (Limbus Technologies GmbH, Rostock, Germany). The final list of called variants in SORD was filtered according to the following criteria: (1) nonsynonymous variants present in a homozygous or compound heterozygous state only, (2) minor allele frequency < 1%, and (3) exon distance of ±30 bp.
PCR and Sanger sequencing of SORD and SORD2P
PCR and Sanger sequencing was used to amplify coding exons and flanking introns of SORD and exon 7 of SORD2P from genomic DNA. Primer sequences, including internal distinct primer, concentrations and PCR thermocycling conditions, were used in analogy to Cortese et al. (2). Sequences were analyzed using ABI 3730XL DNA analyzer (Applied Biosystems, Waltham, MA, USA) in accordance with the manufacturer’s protocol.
Results
Identification of biallelic SORD variants in undiagnosed neuropathies
Five patients in our cohort were found to harbor pathogenic variants in SORD (3%). Four cases were found to carry the most common pathogenic frameshift variant c.757delG (p.Ala253Glnfs*27) in exon seven of SORD in a homozygous state (ClinVar # 929258). One case carried the previously described variant and an additional pathogenic missense variant c.458C > A (p.Ala153Asp) (ClinVar # 1012846) in exon five in a compound heterozygous state. The locations and distributions of SORD variants in previous studies and our study are presented in and (Cortese et al., Citation2020; Li et al., Citation2023; Liu et al., Citation2021; Pons et al., Citation2023; Yuan et al., Citation2021).
Figure 1. (A) Overview of pathogenic SORD variants. Schematic representation showing all exons (green boxes) and introns (blue boxes) based on the NCBI reference sequence NM_003104.6. The SORD variants reported in patients with CMT in this study (top) and mutations described already in the literature (bottom) map throughout the coding region of the gene. (B) SORD protein ortholog alignments showing that the missense substitution p.Ala153Asp of the reported patient V in this article is located at highly conserved residues across species.
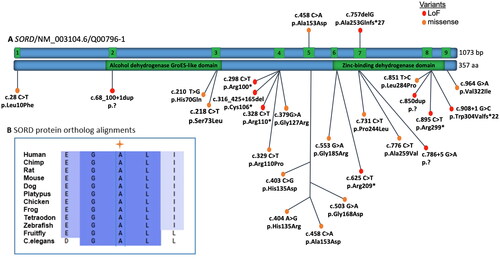
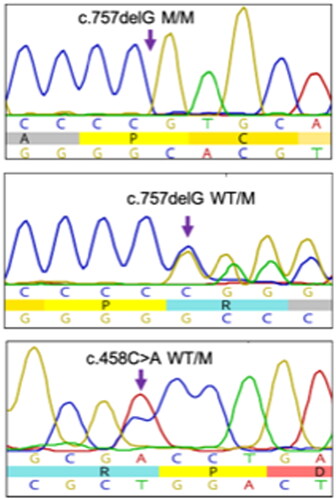
Figure 2. Representative electropherograms show that the c.757delG (p.Ala253Glnfs*27) variant is found in a homozygous and the c.757delG (p.Ala253Glnfs*27) and c.458C > A (p.Ala153Asp) in a compound heterozygous state in patients with CMT from our cohort.
Clinical features of patients with SORDD neuropathy
Clinical and electrophysiological findings comprised signs of slowly progressive, predominantly distal weakness with leading axonal neuropathy in the majority of identified cases. Age of onset ranged from early infancy to mid-twenties. There was no family history of neuromuscular disease apart from one potentially affected sister of Patient V. Neither sister nor parents of Patient V were available for genetic testing and clinical examination. All patients were of German ancestry apart from Patient II who is of Turkish descent with consanguineous background. No patient underwent nerve biopsy prior to genetic diagnosis. Extensive diagnostic work-up including laboratory and CSF analyses remained normal in all patients. NGS panel testing for inherited neuropathies performed in all patients prior to discovery of SORD revealed no pathogenic variants.
Detailed clinical and electrophysiological findings of all patients are summarized in and , respectively.
Table 1. Clinical findings in SORDD patients.
Table 2. Electrophysiology results in SORDD patients.
Discussion
We have confirmed pathogenic variants in SORD as causative for a small subset of prior genetically unconfirmed axonal CMT and dHMN in our cohort from a large neuromuscular center in Northern Germany.
Frequency studies
The detection rate for biallelic pathogenic variants in SORD was 3% (5/166) in our study. These results were overall consistent with those from previous studies with frequencies ranging from 1.4% to 13.8% depending on inclusion criteria (CMT, intermediate CMT, CMT2, dHMN; Cortese et al., Citation2020; Dong et al., Citation2021; Liu et al., Citation2021; Pons et al., Citation2023; Yuan et al., Citation2021). The original study of Cortese et al. (Citation2020) identified up to 10% SORD-related neuropathies in an international cohort of undiagnosed dHMN and CMT2 cases. Various Chinese cohorts have been reporting frequencies ranging from 1.4% to 13.8% (Dong et al., Citation2021; Liu et al., Citation2021; Yuan et al., Citation2021). More recently, there have been cohorts published from Czech, French, and Swiss centers with similar frequencies (Laššuthová et al., Citation2021; Pons et al., Citation2023); to our best knowledge, our report currently gives the first frequency estimation for a German cohort stemming from a large Northern German neuromuscular center.
Clinical findings
Clinical features presented here for the German cohort are consistent with those from previous studies. Age of onset ranged from early infancy to mid-twenties in our cohort as reported before. Clinical findings comprised signs of slowly progressive and predominantly distal weakness and muscle wasting in all cases. Four cases displayed electrophysiological signs of axonal CMT and one case of dHMN without sensory impairment and with normal SNAP strengthening the importance of recognizing early-onset, slowly progressive axonal neuropathy, but also distal motor forms as important red flags triggering diagnostic screening for SORDD.
Animal models and clinical trial using aldose reductase inhibitors
Serum sorbitol dosage is an established functional test for detection of impairment of the enzyme sorbitol dehydrogenase. Pons et al. used serum sorbitol dosages to classify SORD variants as pathogenic and discussed high serum sorbitol level as a potential biomarker to monitor future therapeutic trials using aldose reductase inhibitors (Pons et al., Citation2023).
Experimental treatment with aldose reductase inhibitors has been shown to normalized intracellular sorbitol levels in patient-derived fibroblasts and in Drosophila melanogaster and ameliorated the identified motor and eye phenotype (Cortese et al., Citation2020). More recently, a Sord-/- rat model has become available showing increased levels of sorbitol in serum, CSF, and tissues, and elevated levels of NfL in serum. Besides, Sord-/- rats develop a motor-predominant neuropathy and delayed motor performance that closely resembles the human phenotype (Rebelo et al., Citation2024). Based on these preclinical studies in human-derived cells, fly models and a rat knockout-model, it will be worthwhile to assess and monitor sorbitol and NfL levels in this cohort to further explore treatment options with pharmacological inhibition of aldose reductase and to fully explore NfL as a marker for progression of axonal degeneration alongside with further clinical and electrophysiological measures.
Conclusion
We have confirmed pathogenic variants in SORD as causative for a small subset of prior genetically unconfirmed CMT 2 and dHMN in our cohort from a large neuromuscular center in Northern Germany. Our findings and recent findings from the literature thus strengthen the concept that screening of the SORD gene needs to be performed in patients with genetically unconfirmed CMT, especially axonal, intermediate, and motor-predominant forms.
Authors’ contributions
AA, EAÖ, AS, SR, BS, and SW analyzed and interpreted the molecular genetic and patient data. AA and SW wrote the manuscript. GMzH and HW supervised the study. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Informed consent was obtained from all patients and the ethics committee of the University Hospital Münster granted ethical approval.
Consent for publication
No identifiable data is present in the manuscript. Patients gave consent to use non-identifiable clinical and genetic routine data for scientific purposes.
Acknowledgments
The authors thank the patients who participated in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Cortese, A., Zhu, Y., Rebelo, A. P., Negri, S., Courel, S., Abreu, L., Bacon, C. J., Bai, Y., Bis-Brewer, D. M., Bugiardini, E., Buglo, E., Danzi, M. C., Feely, S. M. E., Athanasiou-Fragkouli, A., Haridy, N. A., Isasi, R., Khan, A., Laurà, M., Magri, S., … Zuchner, S. (2020). Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nature Genetics, 52(5), 473–481. https://doi.org/10.1038/s41588-020-0615-4
- Dong, H.-L., Li, J.-Q., Liu, G.-L., Yu, H., & Wu, Z.-Y. (2021). Biallelic SORD pathogenic variants cause Chinese patients with distal hereditary motor neuropathy. NPJ Genomic Medicine, 6(1), 1. https://doi.org/10.1038/s41525-020-00165-6
- Gabbay, K. H. (1973). The sorbitol pathway and the complications of diabetes. The New England Journal of Medicine, 288(16), 831–836. https://doi.org/10.1056/NEJM197304192881609
- Jedziniak, J. A., Chylack, L. T., Jr., Cheng, H. M., Gillis, M. K., Kalustian, A. A., & Tung, W. H. (1981). The sorbitol pathway in the human lens: Aldose reductase and polyol dehydrogenase. Investigative Ophthalmology & Visual Science, 20(3), 314–326. https://www.ncbi.nlm.nih.gov/pubmed/6782033
- Kato, N., Yashima, S., Suzuki, T., Nakayama, Y., & Jomori, T. (2003). Long-term treatment with fidarestat suppresses the development of diabetic retinopathy in STZ-induced diabetic rats. Journal of Diabetes and Its Complications, 17(6), 374–379. https://doi.org/10.1016/s1056-8727(02)00193-9
- Laššuthová, P., Mazanec, R., Staněk, D., Sedláčková, L., Plevová, B., Haberlová, J., & Seeman, P. (2021). Biallelic variants in the SORD gene are one of the most common causes of hereditary neuropathy among Czech patients. Scientific Reports, 11(1), 8443. https://doi.org/10.1038/s41598-021-86857-0
- Li, L., Xie, Y., Zeng, S., Li, X., Lin, Z., Huang, S., Zhao, H., Cao, W., Liu, L., Liu, J., Rong, P., & Zhang, R. (2023). Expanding the genetic and clinical spectrum of SORD-related peripheral neuropathy by reporting a novel variant c.210T > G and evidence of subclinical muscle involvement. Journal of the Peripheral Nervous System, 28(4), 608–613. https://doi.org/10.1111/jns.12591
- Liu, X., He, J., Yilihamu, M., Duan, X., & Fan, D. (2021). Clinical and genetic features of biallelic mutations in SORD in a series of Chinese patients with Charcot-Marie-Tooth and distal hereditary motor neuropathy. Frontiers in Neurology, 12, 733926. https://doi.org/10.3389/fneur.2021.733926
- Oates, P. J. (2002). Polyol pathway and diabetic peripheral neuropathy. International Review of Neurobiology, 50, 325–392. https://doi.org/10.1016/s0074-7742(02)50082-9
- Oates, P. J., & Mylari, B. L. (1999). Aldose reductase inhibitors: Therapeutic implications for diabetic complications. Expert Opinion on Investigational Drugs, 8(12), 2095–2119. https://doi.org/10.1517/13543784.8.12.2095
- Obrosova, I. G., Van Huysen, C., Fathallah, L., Cao, X., Greene, D. A., & Stevens, M. J. (2002). An aldose reductase inhibitor reverses early diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. Federation of American Societies for Experimental Biology. The FASEB Journal express article 10.1096/fj.01-0603fje. https://deepblue.lib.umich.edu/bitstream/handle/2027.42/154366/fsb2fj010603fje.pdf?sequence=2
- Pons, N., Fernández-Eulate, G., Pegat, A., Théaudin, M., Guieu, R., Ripellino, P., Devedjian, M., Mace, P., Masingue, M., Léonard-Louis, S., Petiot, P., Roche, P., Bernard, E., Bouhour, F., Good, J.-M., Verschueren, A., Grapperon, A.-M., Salort, E., Grosset, A., … Bonello-Palot, N. (2023). SORD-related peripheral neuropathy in a French and Swiss cohort: Clinical features, genetic analyses, and sorbitol dosages. European Journal of Neurology, 30(7), 2001–2011. https://doi.org/10.1111/ene.15793
- Rebelo, A. P., Abad, C., Dohrn, M. F., Li, J. J., Tieu, E. K., Medina, J., Yanick, C., Huang, J., Zotter, B., Young, J. I., Saporta, M., Scherer, S. S., Walz, K., & Zuchner, S. (2024). SORD-deficient rats develop a motor-predominant peripheral neuropathy unveiling novel pathophysiological insights. Brain, Mar 27:awae079. https://doi.org/10.1093/brain/awae079
- Rossor, A., Polke, J., Houlden H., Mary M Reilly M. (2013). Clinical implications of genetic advances in Charcot-Marie-Tooth disease (2013). Nat Rev Neurol (10), 562–71. https://doi.org/10.1038/nrneurol.2013.179.
- Yuan, R.-Y., Ye, Z.-L., Zhang, X.-R., Xu, L.-Q., & He, J. (2021). Evaluation of SORD mutations as a novel cause of Charcot-Marie-Tooth disease. Annals of Clinical and Translational Neurology, 8(1), 266–270. https://doi.org/10.1002/acn3.51268