
Abstract
Purpose: Alveolar epithelium dysfunction is associated with a very large spectrum of disease and an abnormal repair capacity of the airway epithelium has been proposed to explain the pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Following epithelium insult, the damaged cells will activate pathways implicated in the repair process, including proliferation and acquisition of migratory capacities to cover the denuded basement membrane. Induction of Endoplasmic Reticulum stress may be implicated in this process. Interestingly, ER stress excessive activation has been proposed as a central event associated with aberrant repair process and cellular dysfunction observed in IPF. Methods: We study by wound healing assay the molecular targets associated with Alveolar Epithelial Cells (AEC) repair. Results: We demonstrate that the wound recovery of AEC is associated with TGF-β1 signaling and increased transcriptional activity of ER stress and HIF-dependent genes. We further demonstrated that inhibition of TGF-β1 signaling, CHOP expression or HIF-1 expression, limits AECs wound closure. Conclusion: the use of pharmacological drugs targeting the ER/HIF-1 axis could be an attractive approach to limit AEC dysregulation in pathological condition, and confirmed a critical role of theses factor in response to alveolar injury.
Keywords:
Introduction
Lung homeostasis is ensured by the perfect integrity and functionality of the alveolar epithelium. This one, composed of type I and type II alveolar epithelial cells (AEC) provides an extensive surface for gas exchange, and represents a physical barrier that protect lungs from environmental insults. Thus, alveolar epithelium dysfunction is associated with a very large spectrum of disease including acute lung injury (ALI) and pulmonary fibrosis.Citation1,Citation2 More specifically, AEC dysfunction and an abnormal repair capacity of the airway epithelium has been proposed to explain the pathogenesis of Idiopathic Pulmonary Fibrosis (IPF).Citation2
A pleiotropic cytokine, the transforming growth factor-β1 (TGF-β1), is the major inducer of alveolar epithelial cells (AECs) response to injury.Citation3 In physiological condition, following epithelium insult, the damaged cells will secrete TGF-β1 which is perceived by the neighboring cells, through specific TGF-β1 receptors, conducting to the activation of pathways implicated in the repair process. These pathways include proliferation and expression of a-SMA protein by AEC cells, in order to acquire for a short reversible time migratory capacitiesCitation4 to completely cover the denuded basement membrane. Recently, some studies pointed out the critical role of endoplasmic reticulum (ER) stress and un-folded protein response (UPR) activation in the migration capacity of cancer cellsCitation5,Citation6 or nasal fibroblastCitation7 in response to TGF-β1 stimulation. Therefore, induction of ER stress may be implicated in the repair process of AECs.Citation8 Induction of the UPR pathways is an adaptive response to loss of ER homeostasis, which consists on the activation of activating transcription factor 4 (ATF4), ATF6α and spliced X-box binding protein 1 (XBP1) transcription factors. These actors are responsible of the inhibition of protein translation, the increased chaperon synthesis and protein folding, the activation of the ER Activated Degradation (ERAD) system to limit formation of proteins aggregates, and cell apoptosis.
Expression of ER stress markers has been observed in AECs from biopsies of IPF patients,Citation9 and proposed as a central event associated with aberrant repair process and cellular dysfunction observed in the course of the disease. Indeed, apoptosis of AEC and loss of epithelial phenotype and acquisition of mesenchymal markers in AECs has been related to ER stress induction.Citation10,Citation11 Interestingly, we and other proposed that expression of ER stress markers was related to a specific hypoxic micro-environment of the cellsCitation12–14 due to tissue remodeling. More, we demonstrate that the main orchestrator of the hypoxic response, HIF-1, is able to trigger ER stress and CHOP-mediated apoptosis independently of the hypoxic status of the cell.Citation12
Here, we study the molecular targets associated with AECs repair. We demonstrate that HIF-1 and ER stress, (especially CHOP) are implicated in the wound closure process after injury. This study strengthens the concept that the HIF-1/ER stress axis is a key event in alveolar response to injury, and especially in the repair capacity of the alveolar epithelium; and that targeting this pathway could be an attractive approach to limit AEC pathological dysregulation as observed in pulmonary fibrogenesis and exacerbation.Citation12
Materials and methods
Primary rat AECs isolation
Experimental protocols were approved by the Ethics Committee for Animal Experiment Charles Darwin (Ce5) and the French ministry of research (APAFIS#8150). Primary AECs were isolated from 4 weeks male Sprague-Dawley rats. Lungs were digested with 60 U elastase (Worthington) and AECs were purified by a differential adherence technique on plastic plates (Greiner, ThermoScientific). AECs were grown in DMEM containing 25 mM D-glucose, 10 mM Hepes, 23.8 mM NaHCO3, 2 mM L-glutamine, 10% fetal bovine serum (FBS), 50 U/ml penicillin, 50 µg/ml streptomycin, 10 µg/ml gentamycin, 10 µg/ml amphotericin B (Thermo Scientific) at 37 °C with 5% CO2 in a humidified incubator. As describe previously, the percentage of AT2 cells as assessed by phosphine 3 R staining of lamellar bodies was 92% of freshly isolated cells, and cell viability was 95%.Citation15 For each experiment, AEC from lungs of at least 3 rats were used.
Primary rat AECs treatments
On the third day after isolation, AECs were treated with pharmacological drugs. 100 mM of 4-phenylbutyric acid (4-PBA) (Sigma Aldrich) was used at to relieve the ER stress, 10 µM of the 1′-hydroxymethyl-2′-furyl)-1-benzyl indazole (YC-1) was used to inhibit HIF-1α stabilization, 10 µM of SB431542 was used to inhibits of TGF-β1 type I receptor kinase (Sigma Aldrich), and 1 ng/ml of recombinant TGF-β1 (R&D Systems, Lille, France) was used to stimulate wound closure. Pharmacological drugs have been added in the culture media at the same time as wound injury.
Wound closure assay
The highly reproducible wound closure techniqueCitation16,Citation17 has been used to estimate the repair capacity of AECs after mechanical injury. On day 3 after isolation, primary AECs grown on labteck were injured mechanically with a 10ul tip in the middle of the plate culture. AECs were washed with PBS to detached injured cells and fresh medium was added in presence or absence of specific inhibitors 4-PBA, YC-1 or SB431542. Pictures were taken immediately after the scratch and 16h after the injury with a Retiga 2000 R CCD camera with Image-proexpress 6.0 (QImaging) connected to an Axio Observer D1 inverted epifluorescence microscope (Zeiss). Marks on the Petri dishes allowed us to photograph the wounds exactly at the same place at various times. Wound closure was determined as a percentage of the area recovered at the end of the experiment as compared to the same area at the beginning. Analysis has been done with Image J software.
Transient transfection of AECs and luciferase activity assay
AECs were transiently transfected with the NEON™ transfection system (Life Technologies) as previously de-scribed.Citation18 The cloned amino acid response element (AARE) inserted upstream of the firefly luciferase reporter gene allow to study the ATF4 transcriptional activity (6.1 kb pGL3-2xAARE)Citation19 and the cloned endoplasmic reticulum response element (ERSE) inserted upstream of the firefly luciferase gene allow to evaluate ATF6/XBP-1s transcriptional activity (pGL4.39 Promega). HIF transactivation capacity was study with an hypoxia response element (HRE) cloned upstream the luciferase gene (pGL4.42 Promega). CHOP promotor activity was studied using CHOP-luc plasmid (a generous gift from Dr Alain BruhatCitation20) and construct as −649 bp of the CHOP promotor cloned into a pGL3-basic reporter construct (Promega). The plasmid pRL-SV40 expressing renilla reniformis luciferase (RL) reporter gene (Promega) was used for normalization of the luciferase response. On day 1 after isolation, AECs electroporation were performed with 1 µg of the plasmid of interest (AARE-luc, ERSE-luc, HRE-luc, CHOP-luc) and with 0.4 µg of pRL-SV40. After electroporation, cells were seeded in 24-wells without antibiotics in supplemented DMEM. On day 3, AECs were exposed to wound closure assay. At the end of exposition, AECs were lysed to perform firefly and RL assays with the Dual-Luciferase Reporter Assay System, as specified by the manufacturer (Promega). AARE, ERSE, HRE and CHOP activities were evaluated by quantifying the relative light units (RLU) ratio of firefly to RL measured with the Clarity™ luminescence microplate reader (BioTek). Activation of each responsive element by overexpression of specific transcription factor has been tested previously.Citation12,Citation13
Immunofluorescence staining
Immunostaining were realized before or after the mechanical injury of AECs cultured at T0 and T16h. After 4% paraformaldehyde fixation and 0,1% Triton X100 permeabilization, AECs were incubated 30 min in 2% BSA blocking solution and 1 h with primary antibody anti-α-SMA (Sigma Aldrich, A2547) and anti-TTF1 (WRAB-1231, Seven Hills Bioreagents). AECs were washed with PBS and incubated 1 hour in 1:200 Alexa 488 and 568 Fluor® conjugated secondary antibodies (Invitrogen). Nuclear labeling was performed by DAPI (300 nM) staining during 8 min and washed with PBS. Slides were mounted with ImmunMount solution (Thermo Scientific).
Determination of TGF-β level
The supernatants of AECs were collected, spun at 12,000 × g for 15 min at 4 °C, and stored at −80 °C for further processing. The concentrations of active TGF-β1 were determined using specific Quantikine ELISA kits (R&D Systems, Lille, France) according to the manufacturer’s instructions.
Gene silencing
As transfection efficiency in primary rat AECs is low (less that 30% of cells expressing the transgene), we used the human alveolar epithelial A549 cell line (ATCC) for gene silencing experiments. A549 cells were cultured in same conditions as primary AEC. A549 cells were transfected with CHOP siRNA sequences 5’AAGAACAGCAGAGGUCACAA-ttt3’, 5’GCCUGGUAUGAGGACCUGC-ttt3’ or with HIF-1 siRNA sequence 5’CUGAUGACCAGCAACUUGA-ttt3’ using Lipofectamine® 2000 according to the manufacturer’s instructions (ThermoFisher). As reported in previous works, transfection efficiency reached 80%.Citation12
Statistical analyses
Statistical analyses were performed using PRISM software (version 9 GraphPad). Results were presented as means ± SD. Shapiro-Wilk and Kolmogorov-Smirnov test were used to assess the distribution of the data. Welsh’s t-test was performed to evaluate differences between treated groups versus untreated groups. An ordinary one way ANOVA with a Sidak post-hoc test was performed to evaluate multiple comparisons. A p value < 0.05 was considered as a significant difference between conditions.
Results
Acquisition of migratory capacity of AECs after mechanical injury and autocrine regulation of wound recovery
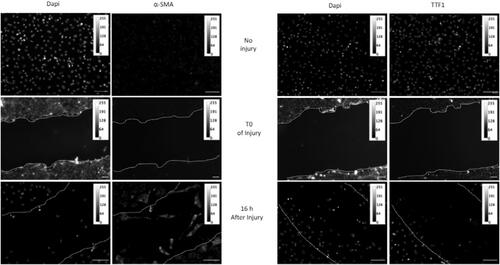
To study the migratory capacity of damaged AECs, we used the highly reproductive model of mechanical injury by a tip scratch of the cell monolayer.Citation21,Citation22 Supplemental Figure 1 shows the evolution of the wound area a T0 of injury and 16h and 24h after scratch, and the reproducibility of the technique. At T0, every AEC present in the dish expressed TTF1 marker, whereas no staining of α -SMA was detected. During the recovery, we observed in wound space AEC with lamellipodia expressing α-SMA (). This mesenchymal phenotype is accompanied by a loss of TTF1 expression, in the migrating cells.
Figure 1. Acquisition of migratory capacity of AECs after mechanical injury. Confluent primary AECs monolayers plated into labteck were mechanically injured by scratch. At T0 and 16 hours later, cells were fixed and immunostaining of α-SMA or TTF1 were performed. Images were presented in 8-bit grey panel. A calibration bar has been added in each picture. A representative picture of at least n = 3 independent AEC cultures for each condition has been presented. Original magnification: × 200, Scale Bar rep-resent 50 µm.
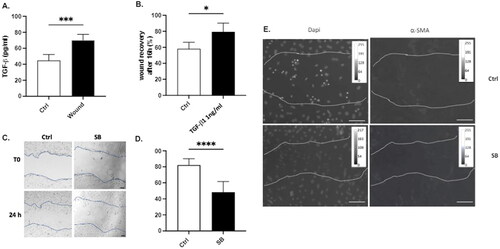
TGF-β1 signaling has been shown to promote repair in AEC after injury.Citation3 In our model, an increase in TGF-β1 secretion is observed 24 hours after cell injury (), and treatment of AECs with 1 ng/ml of TGF-β1 accelerates wound healing (). Indeed, while 58,2% of the area is recovered in control condition at 16h post injury, 79,4% has been recovered in cells treated with TGF-β1 (). More, we observed that, whereas 82.2% of the wound is recovered at 24h post injury, only a 48.24% is observed when cells were treated with SB431542, a previously validate inhibitor of TGF-β1 type I receptor kinase () limiting AEC Epithelial to Mesenchymal Transition.Citation10,Citation15 Finally, we demonstrate that treatment of AEC with SB431542 also limits the expression of α-SMA on migrating cells 16h after injury ().
Figure 2. Implication of TGF-β1 in the wound healing capacity of AECs. (A) Confluent primary AECs were mechanically injured. 24h later, TGF-β1 release was evaluated by ELISA in the supernatant. (B-D) Confluent primary AECs monolayer were mechanical-ly injured, and treated either with 1 ng/ml of recombinant TGF-β1 (B) or with 10 µM SB431542 (SB), an inhibitor of TGF-β1 type 1 receptor kinase (C-D). 16 hours (B) or 24 hours (C-D) later, cells were fixed and wound closure was determined as a percentage of the area recovered at the end of the experiment as compared to the same area at the beginning. Analysis has been done with Image J software. Raw data were submitted a t test with Welsh correction. At least n = 5 independent experiments from lungs of n = 3 distinct rats have been done for each condition. A representative picture has been presented. * indicates a significant difference as compared with control value with p < 0.05, ** p < 0.005, *** p < 0.001. Original magnification: × 200. Scale bar represent 15 µm. (E) Cells were injured and treated or not with 10 µM SB431542 (SB). 16 hours after injury, cells were fixed and immunostaining of α-SMA were performed. Images were presented in 8-bit grey panel. A calibration bar has been added in each picture. A representative picture has been presented. Original magnification: × 200, Scale Bar rep-resent 50 µm.
HIF-1α and ER stress are implicated in the wound closure and TGF-β1 secretion of injured AECs
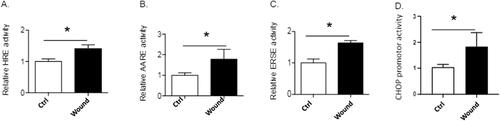
Mechanical injury of AECs layer led to the induction of both the HIF and the UPR signaling pathways. Indeed, the luciferase activities of the HIF (), ATF4 and ATF6N/sXBP1 () responsive elements (HRE, AARE and ERSE respectively) as well as the CHOP promoter () were increased in injured condition.
Figure 3. Activation of HIF and UPR signaling pathways. Primary AECs transfected with plasmid coding for luciferase reporter activity of hypoxia responsive element (HRE: i.e., HIF), amino acid response element (AARE: i.e., ATF4-luc) endoplasmic reticulum stress element (ERSE: i.e., ATF6N/sXBP1-luc), or CHOP promotor were mechanically injured or not and cultured for 24h. Luciferase activity corresponding to the transcriptional capacity of HIF (A), ATF4 (B), ATF6N (C) or activation of CHOP promotor (D) was measured. Independent experiments from lungs of n = 3 distinct rats have been done for each condition. Raw data were submitted to a Welsh’s t-test. * indicates a significant difference as compared with control value with p < 0.05.
Silencing of HIF1-α and CHOP before mechanically injure significantly reduced wound closure of alveolar cell line A549 after 24 hours (). In the same way, AECs treated with either HIF-1 or ER stress inhibitors (YC-1 and 4-PBA respectively) showed much slower wound closure as compared with untreated cells in which the wound was completely covered after 24 h. Treatment of AECs with 4-PBA reduced the wound closure to 44.36% and YC-1 to 41.78% ().
Figure 4. Inhibition of ER stress and HIF-1α activation limits wound closure and TGF-β1 secretion. (A) A549 cells were transfected with scrambled (sc) siRNA, CHOP siRNA and HIF-1α siRNA. Confluent monolayers were then mechanically injured. 24 hours later, cells were fixed and wound closure was determined. (B) Confluent primary AECs monolayer were mechanically injured and treated with either 100 mM of the ER stress inhibitor 4-PBA or 10 µM of the HIF inhibitor YC-1. 24 hours later, cells were fixed and wound closure was determined. (A-B) Wound closure was determined as a percentage of the area recovered at the end of the experiment as compared to the same area at the beginning. Analyses have been done with Image J software. (C) Confluent primary AECs monolayer were mechanically injured and treated with either 100 mM of 4-PBA or 10 µM of YC-1. 24h later, TGF-β1 release was evaluated by ELISA in the supernatant. Raw data were submitted to an ordinary one way ANOVA with Sidak’s multiple comparison test. At least n = 5 independent experiments from lungs of n = 3 distinct rats have been done for each condition. A representative picture has been presented. * indicates a significant difference as compared with control value, # indicates a significant difference as compared with wound value. * or # indicates a p < 0.05; ** or ## indicates a p < 0.005, *** or ### indicate a p < 0.001.
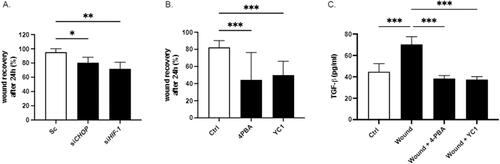
Finally, we demonstrate that pharmacological treatment of injured AEC with HIF-1 and ER stress inhibitors YC-1 and 4-PBA respectively also limits TGF-β1 secretion ().
Discussion
AEC dysregulation and impairment of the alveolar epithelium repair process are currently recognized as one of the major cause of idiopathic pulmonary fibrosis (IPF). Many molecular and cellular mechanisms are involved in the process of re-epithelialization. Among them, the migration of epithelial cells and the reversible acquisition of migratory properties seems to be a major event. Independently of each other, TGF-β1, HIF-1α, and UPR signaling pathways which are strongly implicated in the fibrogenic process, have also been shown to promote wound closure.
In this study, we first demonstrate that after mechanical injury of the monolayer AEC are able to promote the expression of mesenchymal markers such as α-SMA over epithelial markers such as TTF1, to migrate within the wound, and to close the gap. This phenomenon, observed in normal wound healing is the consequence of the induction of the type II Epithelial to Mesenchymal Transition (EMT) process.Citation23 However, if abnormal, it may play a fundamental role in excessive tissue repair as seen in IPF. Basically, this would explain the observed presence of epithelial cells expressing mesenchymal markers in IPF lung biopsies.Citation24,Citation25 Further studies are now necessary to characterized the molecular mechanism of epithelial to mesenchymal transition after repetitive epithelial wound injuries.
TGF-β1 has been shown to drive the EMT process and to promote physiological repair. However, if excessive, TGF-β1 may also activate pathological signaling pathways implicated into the establishment of progressive fibrosis.Citation26 In our study, TGF-β1 accelerate wound closure, and the use of TGF-β1receptor inhibitor delayed this response.
ER is particularly active during wound healing, due to the high rate protein synthesis and folding needed to close the scar.Citation27 More, activation of UPR has been reported as an important driver of TGF-β1-induced α-SMA and collagen type I expression,Citation28,Citation29 and in the induction of EMT in lung epithelial cells,Citation30,Citation31 promoting pulmonary fibrosis.Citation13,Citation32 In this study, we try to demonstrate that that TGF-β1 effects on wound closure were linked to transcriptional activity of ER stress and HIF-dependent genes. Although the experimental procedure of injury does not allow to observe a modification of the intracellular levels of the different targets (data not shown), we observed in our conditions, the transcriptional activation of ATF4 and ATF6/XBP-1 responsive genes, and the induction of CHOP promotor. Moreover, the silencing of CHOP, as well as the use the chemical chaperone 4-PBA, reduce wound recovery. These observations, in accordance with other studies demonstrating that treatment with 4-PBA downregulates TGF-β1-induced migration and collagen contractionCitation7 or implicating ER stress markers in TGF-β1-induced pro-fibrotic response and the promotion of myofibrotic differentiation of lung fibroblastsCitation33 in IPF, reinforce the key role of the activation of ER stress and CHOP in the repair process after wound injury in AEC.
In parallel, activation of the transcriptional factor HIF-1α has been shown to promote proliferation and spreading during the repair process after injury or hypoxic-like condition.Citation34 Interestingly, HIF-1α also play a pivotal role during the process of wound healing independently of hypoxic status of the cells.Citation35–37 In our experiment, we notice the activation of the HIF-Responsive Element in injured AEC, and we show that silencing HIF-1α by siRNA or pharmacological inhibition significantly reduce the wound recovery independently of a hypoxic exposition of AEC. Although our results suggest the involvement of HIF-1α in AEC wound healing, rescue experiments would strengthen this postulate.
In this study, we investigated the role of HIF-1α or the UPR on TGF-β1 release after mechanical injury. Indeed, a pro-fibrotic synergy for HIF-1α and TGF-β1 signaling has been proposedCitation38 since HIF-1α induced TGF-β1 released and conversely, TGF-β1 could increase HIF-1α signaling. Moreover, we recently have proposed a relationship between HIF-1α and ER stress,Citation12,Citation13 as key event in apoptosis and EMT in pulmonary fibrosis. Here, we demonstrate that in response to scratch injury, pharmacological inhibition of HIF-1α or the UPR pathway abrogates TGF-β1 secretion and delay the wound closure. This observation support recent publication reporting an inhibitory effect of 4-PBA on TGF-β1 secretion in lung epithelial cells, and an impact of ER stress inhibition on EMT.Citation39
Conclusions
From the results of this study, we can conclude that AEC wound healing is achieved through extracellular signalization of TGF-β1. These effects involved the transcriptional activity of HIF-1α and the UPR effective transcription factors ATF4, ATF6 and CHOP. Thus, and based on previous observations of our group,Citation12,Citation13 we proposed that repeated and persistent micro-injuries of the airway epithelium could chronically induce activation of HIF-1α and CHOP transcription factors to repair damaged cells. This activation could amplify TGF-β1 release, and participate in the dysregulation of AEC,Citation12,Citation40,Citation41 and pulmonary fibrogenesis.
Author contributions
Conceptualization, E.B, E.D, N.V.; methodology and validation: E.B, E.D, N.V.; writing—review and editing, E.B, N.V.; supervision, E.B; funding acquisition, EB.
Institutional review board statement
Experimental protocols were approved by the Ethics Committee for Animal Experiment Charles Darwin (Ce5) and the French ministry of research (APAFIS#8150), done in accordance with the European Communities Council Directive of September 22, 2010 (2010/63/EU) for animal care, and conducted in accordance with the French legislation for animal care.
| Abbreviations: | ||
| 4-PBA | = | 4-PhenylButyrAte |
| AARE | = | Amino-Acid Responsive Element |
| AEC | = | Alveolar Epithelial Cell |
| ALI | = | Acute Lung Injury |
| ATF | = | Activating Transcription Factor |
| CHOP | = | C/EBP Homologus Protein |
| ER | = | Endoplasmic Reticulum |
| ERSE | = | ER Stress Element |
| HIF | = | Hypoxia Inducible Factor |
| HRE | = | Hypoxia Responsive Element |
| IPF | = | Idiopathic Pulmonary Fibrosis |
| RL | = | Renilla reniformis Luciferase |
| TTF1 | = | Thyroid Transcription Factor 1 |
| TGF | = | Transforming Growth Factor |
| UPR | = | Unfolded Protein Response |
| XBP-1 | = | X-box Binding Protein 1 |
| YC-1 | = | 5′-hydroxymethyl-2′-furyl)-1-benzyl indazole |
| α-SMA | = | a-Smooth Muscle Actin |
Supplemental Material
Download Zip (545.3 KB)Acknowledgments
The authors would like to thank Pr Carole Planes for helpful discussion and Dr Alain Bruhat for generous gift of pGL3-2xAARE and pCHOP-luc plasmids.
Declaration of interest
The authors declare no conflict of interest.
Additional information
Funding
References
- Geiser T. Mechanisms of alveolar epithelial repair in acute lung injury–a translational approach. Swiss Med Wkly. 2003;133(43–44):586–590.
- Selman M, King TE, Pardo A. Idiopathic pulmonary fibrosis: prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann Intern Med. 2001;134(2):136–151. doi:10.7326/0003-4819-134-2-200101160-00015.
- Buckley S, Shi W, Barsky L, Warburton D. TGF-β signaling promotes survival and repair in rat alveolar epithelial type 2 cells during recovery after hyperoxic injury. Am J Physiol-Lung Cell Mol Physiol. 2008;294(4):L739–L748. doi:10.1152/ajplung.00294.2007.
- Chen J, Li H, SundarRaj N, Wang JH-C. Alpha-smooth muscle actin expression enhances cell traction force. Cell Motil Cytoskeleton. 2007;64(4):248–257. doi:10.1002/cm.20178.
- Linxweiler M, Schorr S, Schäuble N, et al. Targeting cell migration and the endoplasmic reticulum stress response with calmodulin antagonists: a clinically tested small molecule phenocopy of SEC62 gene silencing in human tumor cells. BMC Cancer. 2013;13(1):574. doi:10.1186/1471-2407-13-574.
- Chen D, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. The oxido-metabolic driver ATF4 enhances temozolamide chemo-resistance in human gliomas. Oncotarget. 2017; 8(31):51164–51176.
- Shin JM, Kang JH, Park JH, Yang HW, Lee HM, Park IH. TGF-β1 activates nasal fibroblasts through the induction of endoplasmic reticulum stress. Biomolecules. 2020;10(6):942. doi:10.3390/biom10060942.
- Mulugeta S, Nureki SI, Beers MF. Lost after translation: insights from pulmonary surfactant for understanding the role of alveolar epithelial dysfunction and cellular quality control in fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2015;309(6):L507–L525. doi:10.1152/ajplung.00139.2015.
- Korfei M, Ruppert C, Mahavadi P, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(8):838–846.
- Willis Liebler JM, Luby-Phelps K, Nicholson AG, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol. 2005;166(5):1321–1332.
- Zhou G, Dada LA, Wu M, et al. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol-Lung Cell Mol Physiol. 2009;297(6):L1120–L1130. doi:10.1152/ajplung.00007.2009.
- Delbrel E, Soumare A, Naguez A, et al. HIF-1α triggers ER stress and CHOP-mediated apoptosis in alveolar epithelial cells, a key event in pulmonary fibrosis. Sci Rep. 2018;8(1):17939. doi:10.1038/s41598-018-36063-2.
- Delbrel E, Uzunhan Y, Soumare A, et al. ER stress is involved in epithelial-to-mesenchymal transition of alveolar epithelial cells exposed to a hypoxic microenvironment. Int J Mol Sci. 2019;20(6):1299
- Burman A, Kropski JA, Calvi CL, et al. Localized hypoxia links ER stress to lung fibrosis through induction of C/EBP homologous protein. JCI Insight. 2018;3(16):e99543. doi:10.1172/jci.insight.99543.
- Uzunhan Y, Bernard O, Marchant D, et al. Mesenchymal stem cells protect from hypoxia-induced alveolar epithelial-mesenchymal transition. Am J Physiol-Lung Cell Mol Physiol. 2016;310(5):L439–L451. doi:10.1152/ajplung.00117.2015.
- Ruffin M, Bilodeau C, Maillé É, et al. Quorum-sensing inhibition abrogates the deleterious impact of Pseudomonas aeruginosa on airway epithelial repair. FASEB J Off Publ Fed Am Soc Exp Biol. 2016;30(9):3011–3025.
- Trinh NTN, Privé A, Maillé E, Noël J, Brochiero E. EGF and K + channel activity control normal and cystic fibrosis bronchial epithelia repair. Am J Physiol Lung Cell Mol Physiol. 2008;295(5):L866–L880. doi:10.1152/ajplung.90224.2008.
- Migneault F, Boncoeur É, Morneau F, Pascariu M, Dagenais A, Berthiaume Y. Cycloheximide and lipopolysaccharide downregulate αENaC mRNA via different mechanisms in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2013;305(10):L747–55. doi:10.1152/ajplung.00023.2013.
- Chaveroux C, Sarcinelli C, Barbet V, et al. Nutrient shortage triggers the hexosamine biosynthetic pathway via the GCN2-ATF4 signalling pathway. Sci Rep. 2016;6:27278. doi:10.1038/srep27278.
- Bruhat A, Jousse C, Carraro V, Reimold AM, Ferrara M, Fafournoux P. Amino acids control mammalian gene transcription: activating transcription factor 2 is essential for the amino acid responsiveness of the CHOP promoter. Mol Cell Biol. 2000;20(19):7192–7204. doi:10.1128/MCB.20.19.7192-7204.2000.
- Borthwick LA, McIlroy EI, Gorowiec MR, et al. Inflammation and epithelial to mesenchymal transition in lung transplant recipients: role in dysregulated epithelial wound repair. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg. 2010;10(3):498–509. doi:10.1111/j.1600-6143.2009.02953.x.
- Wettstein G, Bellaye PS, Kolb M, et al. Inhibition of HSP27 blocks fibrosis development and EMT features by promoting Snail degradation. FASEB J Off Publ Fed Am Soc Exp Biol. 2013;27(4):1549–1560.
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi:10.1172/JCI39104.
- Salton F, Volpe MC, Confalonieri M. Epithelial–mesenchymal transition in the pathogenesis of idiopathic pulmonary fibrosis. Medicina (Mex). 2019; 55(4):83. doi:10.3390/medicina55040083.
- Xu Y, Mizuno T, Sridharan A, et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2016;1(20):e90558.
- Lee CM, Park JW, Cho WK, et al. Modifiers of TGF-β1 effector function as novel therapeutic targets of pulmonary fibrosis. Korean J Intern Med. 2014;29(3):281–290. doi:10.3904/kjim.2014.29.3.281.
- Bachar-Wikstrom E, Manchanda M, Bansal R, et al. Endoplasmic reticulum stress in human chronic wound healing: Rescue by 4-phenylbutyrate. Int Wound J. 2021;18(1):49–61. doi:10.1111/iwj.13525.
- Ghavami S, Yeganeh B, Zeki AA, et al. Autophagy and the unfolded protein response promote profibrotic effects of TGF-β1 in human lung fibroblasts. Am J Physiol – Lung Cell Mol Physiol. 2018;314(3):L493–L504. doi:10.1152/ajplung.00372.2017.
- Sharma P, Alizadeh J, Juarez M, et al. Autophagy, apoptosis, the unfolded protein response, and lung function in idiopathic pulmonary fibrosis. Cells. 2021;10(7):1642. doi:10.3390/cells10071642.
- Tanjore H, Cheng DS, Degryse AL, et al. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. 2011;286(35):30972–30980. doi:10.1074/jbc.M110.181164.
- Zhong Q, Zhou B, Ann DK, et al. Role of endoplasmic reticulum stress in epithelial-mesenchymal transition of alveolar epithelial cells: effects of misfolded surfactant protein. Am J Respir Cell Mol Biol. 2011;45(3):498–509. doi:10.1165/rcmb.2010-0347OC.
- Meng X, Liu K, Xie H, et al. Endoplasmic reticulum stress promotes epithelial‑mesenchymal transition via the PERK signaling pathway in paraquat‑induced pulmonary fibrosis. Mol Med Rep. 2021;24(1):525. doi:10.3892/mmr.2021.12164.
- Baek HA, Kim DS, Park HS, et al. Involvement of endoplasmic reticulum stress in myofibroblastic differentiation of lung fibroblasts. Am J Respir Cell Mol Biol. 2012;46(6):731–739. doi:10.1165/rcmb.2011-0121OC.
- McClendon J, Jansing NL, Redente EF, et al. Hypoxia-inducible factor 1α signaling promotes repair of the alveolar epithelium after acute lung injury. Am J Pathol. 2017;187(8):1772–1786. doi:10.1016/j.ajpath.2017.04.012.
- Hou Z, Nie C, Si Z, Ma Y. Deferoxamine enhances neovascularization and accelerates wound healing in diabetic rats via the accumulation of hypoxia-inducible factor-1α. Diabetes Res Clin Pract. 2013;101(1):62–71. doi:10.1016/j.diabres.2013.04.012.
- Zhu Y, Wang Y, Jia Y, Xu J, Chai Y. Roxadustat promotes angiogenesis through HIF-1α/VEGF/VEGFR2 signaling and accelerates cutaneous wound healing in diabetic rats. Wound Repair Regen off Publ Wound Heal Soc Eur Tissue Repair Soc. 2019;27(4):324–334.
- Andrikopoulou E, Zhang X, Sebastian R, et al. Current Insights into the role of HIF-1 in cutaneous wound healing. CMM. 2011;11(3):218–235. doi:10.2174/156652411795243414.
- Qian F, He M, Duan W, et al. Cross regulation between hypoxia-inducible transcription factor-1α (HIF-1α) and transforming growth factor (TGF)-ß1 mediates nickel oxide nanoparticles (NiONPs)-induced pulmonary fibrosis. Am J Transl Res. 2015;7(11):2364–2378.
- Liu D, Zhu H, Gong L, et al. Histone deacetylases promote ER stress induced epithelial mesenchymal transition in human lung epithelial cells. Cell Physiol Biochem. 2018;46(5):1821–1834. doi:10.1159/000489367.
- Weng T, Poth JM, Karmouty-Quintana H, et al. Hypoxia-induced deoxycytidine kinase contributes to epithelial proliferation in pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190(12):1402–1412. doi:10.1164/rccm.201404-0744OC.
- Darby IA, Hewitson TD. Hypoxia in tissue repair and fibrosis. Cell Tissue Res. 2016;365(3):553–562. doi:10.1007/s00441-016-2461-3.