Abstract
Kufs disease, the late-onset form of a group of neurodegenerative disorders, known as the neuronal ceroid-lipofuscinoses, is characterized by intraneuronal/extraneuronal accumulation of proteins that are visible as fingerprint inclusions and granular osmiophilic deposits (GRODs) at the ultrastructural level. A problematic case of Kufs disease in a 53-year-old female affected by progressive gait disturbances, myoclonus, epilepsy, and profound dementia is presented. Laboratory, biochemical, and molecular genetic tests were unremarkable. A magnetic resonance imaging of the brain revealed a moderate atrophy over both hemispheres with no white matter changes. Ultrastructural examination of rectal mucosa biopsies showed fingerprint inclusions in perivascular smooth muscle cells. Pathological examination of autoptic tissues showed numerous intraneuronal PAS-positive, diastase-resistant inclusions corresponding to GRODs at the ultrastructural examination, but no fingerprint inclusions. Cerebellum, skeletal, and cardiac muscles, skin, and liver were unaffected. The present case illustrates the diagnostic difficulties encountered while examining Kufs disease pathological samples. Main problematic issues include (1) specificity and diagnostic value of fingerprint inclusions when found exclusively in perivascular smooth muscle cells; (2) safe distinction of GRODs from lipofuscin inclusions in cerebral tissue; and (3) reliability in using extraneural tissues and, in particular, rectal mucosa biopsies for diagnostic purposes.
The neuronal ceroid–lipofuscinoses (NCLs) collectively constitute the most common type of autosomal recessive inherited neuro-degenerative diseases in childhood [Citation[1]]. However, a rare, late adult onset form, namely Kufs disease, is also described [Citation[1]]. Clinically, these neuro-degenerative disorders are characterized by progressive cognitive decline, blindness, and myoclonic epilepsy, while the rare Kufs disease is dominated by dementia. All forms of NCLs are characterized by intraneuronal accumulation of proteins [Citation[1]], which can be seen as autofluorescent, diastase-resistant periodic acid–Schiff (PAS), and Sudan black B-positive cytoplasmic lipopigment granules at light microscopy. With electron microscopy it is possible to distinguish 5 distinct patterns of osmiophilic lipopigments: lipofuscin, fingerprint deposits, curvilinear bodies, granular osmiophilic deposits (GRODs), and microtubular parallel arrays [Citation[2]].
On the bases of the age of onset, clinical symptoms, and the ultrastructure of the storage material, Dyken identified 4 main forms of NCLs: infantile, late infantile, juvenile, and adult [Citation[3]]. Recent advances in both genetics and biochemistry have led to the identification of 8 distinct NCL subtypes: CLN 1–8 [Citation[4]]. A detailed view of Dyken's classification and currently used genetic classification of NCLs can be found in Boldrini et al. [Citation[2]]. Kufs disease—CLN 4—is the most elusive of the group because, to date, no loci for this NCL subtype have been identified and consequently the diagnosis of the suspicious clinical cases still relies on the morphological examination of affected tissues and the careful exclusion of other possible alternatives.
Histopathological diagnosis of Kufs disease is based on the intraneuronal presence of PAS and Sudan B-positive lipopigment inclusions, which ultrastructurally correspond to unique fingerprint inclusions and/or to, more subtle, GRODs [Citation[5]]. In the reported cases, the lipopigment inclusions have been described in the cytoplasm of neurons and, to a lesser extent, of other cell types including smooth muscle cells, Schwann cells, and eccrine sweat gland epithelial cells [Citation[1], Citation[5], Citation[6]].
Extraneural tissues have been proposed as an alterative to brain biopsy to facilitate diagnostic tissue collection in a number of neurodegenerative lysosomal and peroxisomal disorders [Citation[7]]. Rectal biopsy is simple to perform, scarcely traumatic, and cost-effective. Apart from the epithelial and endocrine components, the rectal biopsy includes numerous cytotypes, e.g., inflammatory cells, vascular cells, smooth muscle cells, Schwann cells, and ganglion cells, which can accumulate specific lipopigment inclusions. In a review on the diagnostic value of extracerebral biopsy in lysosomal and peroxisomal disorders, the diagnostic contribution of rectal biopsy to Kufs disease has been rated as essential [Citation[7]].
Here we report a problematic case of Kufs disease with clinical, histological, and ultrastructural documentation serving as a teaching case to illustrate the many diagnostic traps that can be encountered while examining cerebral and extracerebral tissues, and in particular rectal mucosa, for diagnostic purposes.
CASE REPORT
M.L. was a 53-year-old right-handed woman from Philippines. She had no personal antecedents of neurological disease or drug intake. The familial history could not be recorded. Three years earlier, at the age of 50 years, she began to present behavioral disturbances, loss of memory, and unsteady gait associated with continuous jerking of arms and legs, which were worse on action. Sensation was normal, as were the tendon jerks, and the plantar responses were flexor. Gait was ataxic and disturbed by myoclonus.
Cognitive examination was grossly abnormal. Ocular examination disclosed bilateral macular dystrophy. Results of general physical examination were normal. Both cognitive impairment and jerks increased in the next years and pharmacoresistant partial motor seizures developed. Routine hematological and biochemical investigations, as well as serum blood gases, ammonia, vitamins E and B12, folate, very-long-chain fatty acids, arylsulfatase A, β-galactosidase, and β-hexosaminidase A and B, were all normal. Urinalysis was normal, even if urinary sediment dolichol estimation was not performed. Cerebral spinal fluid examination revealed a normal glucose and protein concentration; oligoclonal banding was absent. Furthermore, testing for the presence of the 14-3-3 protein was negative.
On molecular genetic investigation, we also ruled out mutations of the genes causing dentato-rubro- pallidoluysian atrophy, Huntington chorea, Friedreich ataxia, spino-cerebellar atrophy (SCA) type 1, 2, 3, 6, and 7, myoclonus epilepsy, and ragged-red fibers syndrome, and all known mutations of the prion protein gene on chromosome 20 related to Creutzfeldt Jacob disease. The genotype of the apoliprotein E alleles in this patient was e3–e4. MRI study revealed moderate atrophy over both hemispheres, with no definite white matter abnormalities. Electroencephalographic recordings showed diffuse background slowing associated with spikes and sharp-wave complexes involving both hemispheres. Intermittent photic stimulation never elicited a photoparoxysmal or photomyoclonic response. Electrical stimulation of either median nerve failed to produce a large cortical somatosensory evoked potential.
In 2 different instances the patient underwent rectoscopy for collecting multiple fragments of rectal mucosa suited for ultrastructural investigation. Six months after our first examination, the patient became bedridden and months later died because of pulmonary infection. A pathological examination was made. At autopsy, mild cerebral atrophy was found and multiorgan tissue sampling was performed.
MATERIALS AND METHODS
Fresh rectal endoscopic biopsies were immediately fixed in 2.5% buffered glutaraldehyde, postfixed in 1% osmium tetroxide, and routinely processed for electron microscopy. Thin sections were stained with uranyl acetate and lead citrate and viewed with a Philips 410 transmission electron microscope.
Autoptic samples were collected from the frontoparietal cortex, cerebellum, cardiac and skeletal muscles, skin, and liver. The samples were routinelly fixed in 10% buffered formalin and processed for light microscopy. Paraffin-embedded sections were stained with hematoxylin and eosin as well as with PAS and PAS/D. Fragments from the frontoparietal cortex and skin were additionally fixed in glutaraldehyde and processed for electron microscopy. For immunohistochemistry, frontoparietal cortex paraffin sections were stained with anti-protein tau (Ab-3, 1:200, NeoMarkers, Fremont CA, USA) and anti-ubiquitin (Abi-1, 1:100, NeoMarkers, Fremont CA, USA) polyclonal antibodies followed by routine ABC method. Immunostains were performed by using appropriate controls.
RESULTS
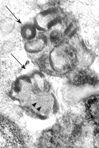
In rectal mucosa biopsies we studied epithelial cells, inflammatory cells, Schwann cells, peripheral nerve endings, endothelial cells, and smooth muscle cells. In one tissue bioptic series, the samples contained only surface elements thus limiting the examination to the rather not diagnostic elements, e.g., epithelial and inflammatory cells. Although extensively searched, no ganglion cells could be identified in all the screened biopsies. Several fingerprint inclusions were located in the cytoplasm of perivascular smooth muscle cells (); no other cytotypes were found to be involved. At high magnification, the inclusions showed the typical association of curvilinear profiles and fingerprint structures (). The latter were composed of stacks of paired, straight or curving, lamellae separated from the adjacent pair by a narrow clear space. No GRODs were seen. These ultrastructural findings were consistent with the diagnosis of Kufs disease.
Fig. 1 Rectal mucosa. Perivascular smooth muscle cell showing fingerprint inclusions (arrows). TEM. Original magnification, × 6,000.
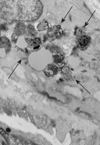
Fig. 2 Rectal mucosa. At high magnification curvilinear profiles (arrowheads) and fingerprints (arrows) are visible. TEM. Original magnification, × 40,000.
Histopathological study of frontoparietal cortex taken at autopsy showed loss of neurons, astrocytosis, and corpora amilacea; numerous intraneuronal PAS-positive, diastase-resistant inclusions were also found (); these inclusions corresponded to GRODs at the ultrastructural examination (). Putative GRODs with a higher lipidic content were also seen (). No fingerprint inclusions were found. Immunohistochemical detection of protein tau and ubiquitine was negative. In the cerebellum no loss of Purkinje cells was found (); the skeletal, cardiac muscles were unaffected; the skin did not reveal any cytoplasmic accumulation of lipopigment in the eccrine sweat gland epithelial cells and fibroblasts both at light and electron microscopy level; the liver showed moderate to severe steatosis.
Fig. 3 Autoptic frontoparietal cortex. PAS-positive diastase-resistant intraneuronal inclusions. PAS/D. Original magnification, × 40.
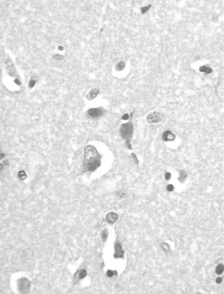
Fig. 4 Autoptic frontoparietal cortex. Collections of granular osmiophilic deposits (GRODs) in the neuronal cytoplasm. TEM. Original magnification, × 6,000.
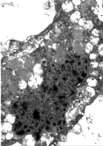
Fig. 5 Autoptic frontoparietal cortex. Detail of putative GRODs associated with neutral lipids. TEM. Original magnification, × 18,000.

Fig. 6 Autoptic cerebellar cortex. The Purkinje cell layer appears well preserved. EE. Original magnification, × 40.
DISCUSSION
The patient described here had a clinical picture consistent with Kufs disease. Although extensively investigated, the laboratory, biochemical, and molecular genetic tests failed to disclose any possible diagnostic alternative. Postmortem autoptic study of the frontoparietal cerebral cortex revealed age-related degenerative lesions as well as PAS-positive, diastase-resistant intraneuronal lipopigment inclusions. Ultrastructurally, these inclusions corresponded to cytoplasmic collections of moderately electron-dense, granular inclusions, which we interpreted as GRODs. According to Carpenter et al. [Citation[8]], GRODs are difficult to distinguish from conventional lipofuscin on electron microscopy. Clues for the GROD nature of the inclusions are uniform matrix granularity, moderate electron density, and poor lipidic content; however, as demonstrates, a clear-cut distinction between such inclusions is not always possible. Consequently, we believe that the final diagnostic interpretation should be driven by the clinical and laboratory background as well as by the absence of any possible diagnostic alternative.
Further, GRODs and fingerprints are not always expressed in the same tissue; in fact, in our patient the fingerprints were found in the rectal mucosa but not in the frontoparietal cortex while the reverse was true with GRODs. This previously unfocused characteristic could, at least in part, explain the plethora of differently textured inclusions that have been published in the past specific literature (for an example, see [Citation[9]]). Moreover, our investigation of extraneural autoptic tissues failed to demonstrate any specific findings; in particular, cutaneous eccrine sweat gland epithelial cells and fibroblasts were unremarkable at both the light and electron microscopy levels. This confirms previous reports that studies of extracerebral tissues give variable results without a consistent diagnostic pattern [Citation[5], Citation[6], Citation[10]].
In the setting of storage diseases, fingerprint inclusions are believed to be virtually unique to Kufs disease. Ultrastructurally, the fingerprint inclusions are composed by tightly bound lamellae that are set in a moderately electron-dense matrix; the lamellae may present various spatial arrangements; whorled (fingerprints), stacked, and prismatic (paracristalline) profiles are possible in relation to the plane of sectioning; the matrix frequently contains curvilinear profiles, also described as C-shaped or worm-like profiles [Citation[11]]. Based on a mandatory, detailed knowledge of the morphological criteria for specific storage, nonspecific structures, and artifacts [Citation[12]], the fingerprint inclusions can be accurately differentiated from conventional lipofuscin bodies, showing coarsely granular, electron-dense material intermixed with large, poorly osmiophilic lipidic globules; multivescicular bodies, showing packed vesicles set in a dense matrix; myelinosomes, containing whorls, stacks of collapsed or reticulated lamellae; and angulated lysosomes of Gaucher-like type, containing sheaves of rigid filaments and lamellae. The present case showed typical fingerprint inclusions in perivascular smooth muscle cells of rectal mucosa, which were consistent with the diagnosis of Kufs disease.
GRODs and fingerprints are believed to be the morphological expression of protein accumulation within cytoplasmic lysosomes, with phospholipids, neutral lipids, and metals accounting for the rest of the inclusion content [Citation[13]]. Mitochondrial ATP synthetase subunit c (SCMAS) is the major protein component of such inclusions at least in 4 types of NCLs (CNL 2, 3, 5, 8) but is not present in infantile NCL (CLN 1), which has been demonstrated to accumulate lysosomal protein saponins (SAP) A&D [Citation[14]]; subunit c has been histochemically documented in CNL 4 and 6 [Citation[15]]; lysosomal protease deficiencies, i.e., palmitoyl protein thioesterase and pepstatin-insensitive peptidase, were demonstrated in CLN 1 and CLN 2, respectively [Citation[13]]. Based on this biochemical setting, it can be suggested that the lipopigment inclusions reflect a general cell protease defect or impairment and that the ultrastructural morphology of these inclusions may amount their biochemical composition, i.e., SAP accumulation corresponding to GRODs and SCMAS to fingerprint/curvilinear inclusions [Citation[1]].
On the practical ground, it is necessary to screen, ultrastructurally, a great diversity of cells even when the inclusions are thought to be restricted to a main cell component. For this purpose, rectal biopsy is simple, safe, scarcely traumatic, and cost-effective; further, many cytotypes can be examined, including the numerous axonal terminals and sustain cells lying free in the lamina propria stroma as well as ganglion cells of mioenteric plexi. However, diagnostic difficulties exist when (1) the biopsy is not deep enough to allow the sampling of ganglion and smooth muscle cells, and this was our experience with the present routine endoscopic material; or (2) a single cytotype presents fingerprint inclusions exclusively. In our case the fingerprints were located in perivascular smooth muscle cells only. As previously discussed, these inclusions have sufficiently distinctive ultrastructural characteristics to be identified with confidence; however, identical structures have been found in the smooth muscle cells of human renal arterioles [Citation[5]], thus raising the possibility that fingerprints could be a previously unrecognized feature of normal smooth muscle cells from human rectal submucosa arterioles.
As illustrated by the present case, the lack of a specific genetic locus alteration makes the diagnosis of Kufs disease rather difficult; in fact, the value of ultrastructural documentation of GRODs and fingerprint inclusions can be narrowed by the paucity of specific findings, monotype cell involvement, dissociated expression of the specific inclusions in tissues, as well as by the variable results observed in the extraneural tissues. To this respect, we suggest that a well-established diagnosis cannot be achieved when only one cytotype is affected by specific inclusions.
REFERENCES
- Haltia M.. The neuronal ceroid-lipofuscinoses. J Nueropathol Exp Neurol. 2003; 62: 1–13
- Boldrini R, Biselli R, Santorelli FM, Bosman C.. Neuronal ceroid lipofuscinosis: an ultrastructural, genetic, and clinical study report. Ultrastruct Pathol. 2001; 25: 51–58. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Dyken PJ.. The neuronal ceroid lipofuscinoses. J Child Neurol. 1989; 4: 165–174. [PUBMED], [INFOTRIEVE]
- Mole SE.. Batten's disease: eight genes and still counting?. Lancet. 1999; 354: 443–445. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Berkovic SF, Carpenter S, Andermann F, Andermann E, Wolfe LS.. Kufs' disease: a critical reappraisal. Brain. 1988; 111: 27–62. [PUBMED], [INFOTRIEVE]
- Goebel HH, Braak H.. Adult neuronal ceroid-lipofuscinosis. Clin Neuropathol. 1989; 8: 109–119. [PUBMED], [INFOTRIEVE]
- Ceuterick-de Groote C, Matin JJ.. Extracerebral biopsy in lysosomal and peroxisomal disorders: ultrastructural findings. Brain Pathol. 1998; 8: 121–132. [PUBMED], [INFOTRIEVE]
- Carpenter S, Karpati G, Wolfe LS, Andermann F.. A type of juvenile cerebromacular degeneration characterised by granular osmiophilic deposits. J Neurol Sci. 1973; 18: 67–87. [PUBMED], [INFOTRIEVE], [CROSSREF]
- Zeman W, Siakotos AN.. Neuronal ceroid-lipofuscinoses, In. Lysosomes and Storage Diseases, Hers HG, Van Hoof F, New York, Academic Press. 1973; 535–542
- Carpenter S.. Morphological diagnosis and misdiagnosis in Batten-Kufs disease. Am J Med Gen. 1988; 5: 85–91
- Ghadially FN.. Ultrastructural Pathology of the Cell and Matrix, ed 4.. London, Butterworth-Heinemann. 1997; 752–755
- Martin JJ, Ceuterik C.. The contribution of pathology to the study of storage disorders. Pathol Res Pract. 1988; 183: 375–385. [PUBMED], [INFOTRIEVE]
- Palmer DN, Fearley IM. Walker JE et al. Mitochondrial ATP synthetase subunit c storage in the ceroid-lipofuscinoses. Am J Med Gen. 1992; 42: 561–567. [CROSSREF]
- Tyynela Y, Suopanky J, Baumann M, Haltia M.. Sphingolipid activator proteins (SAPs) in neuronal ceroid-lipofuscinosis. Neuropediatrics. 1997; 28: 49–52. [PUBMED], [INFOTRIEVE], [CSA]
- Jolly RD, Bown S, Das AM, Walkley SU.. Mitochondrial dysfunction in the neuronal ceroid-lipofuscinosis (Batten disease). Neurochem Int. 2002; 40: 565–571. [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]