Abstract
Heat shock proteins (HSPs) were first identified as stress proteins that confer resistance to physical stresses such as elevated temperatures in all cellular organisms. HSPs are rapidly elevated after stress and confer a temperature resistant phenotype. Temperature resistance is dependent on the ability of HSPs to function as molecular chaperones and prevent aggregation and on the capacity of Hsp27 and Hsp70 to act as wide spectrum inhibitors of the cell death pathways. HSP expression becomes deregulated in cancer leading to elevated expression. Elevated HSP expression promotes cancer by inhibiting programmed cell death (Hsp27, Hsp70) and by promoting autonomous growth (Hsp90) and leads to resistance to chemotherapy and hyperthermia. Tumor HSPs have another property that can be exploited in therapy. They are immunogenic and can be used to form the basis of anticancer vaccines. Elevation in HSP levels may thus have competing effects in tumor growth, being required for tumor cell survival but conferring a hazard for cancer cells due to their immunogenic properties. This dichotomy is also reflected by the approaches used to target HSP in therapy. Pharmacological approaches are being employed to inhibit activity or expression of tumor HSP. Immunological approaches aim at increasing HSP levels in cells and tissues with the aim of increasing tumor antigen presentation to the immune system.
Introduction
With the recent finding that the human genome is unexpectedly compact–with far fewer than the anticipated 100 000 genes, it would appear that genes may be required to encode multiple protein isoforms and proteins may be called on to carry out more than one function. Indeed, globular proteins likely display a number of interaction domains on their surface which are potential interaction or catalytic sites, and it may be that the names given to such proteins only reflect the functions attributed when they were first discovered Citation[1]. Such versatile properties are becoming evident to those of us who investigate the functions of heat shock proteins.
Study of HSP functions was initiated by the finding of a new gene expression pattern, through a happy accident involving the overheating of a Drosophila salivary gland preparation on a microscope stage Citation[2]. This was first reported as: ‘A new puffing pattern induced by temperature shock and DNP in Drosophila’ in 1962 Citation[2]. However, it took another 10–15 years before the first Drosophila HSP mRNA were isolated Citation[3]. Around this time however, (1978) HSPs were discovered in mammalian tissue culture cells, in Escherichia coli, in yeast, and in plants Citation[4–9]. The heat shock field emerged as a major study area in experimental biology at the 1982 meeting ‘Heat Shock: From Bacteria To Man’ held at the Cold Spring Harbor Laboratory Citation[3]. At this time however, the functions of the HSP remained mysterious and the details of regulation of HSP gene expression were only beginning to emerge. All that was known was that the proteins possess ‘homeostatic activity’ and are to this day associated with resistance to heat shock and other stresses. However, in the early 1980s, the concept began to emerge that the HSP belonged to a new kind of protein which functions to modify the structure of other proteins. Most notably the functions of the E coli genes DNA-K (Hsp70), DNA-J (Hsp40) and GroEL (Hsp60) were determine genetically in a study of λ phage replication and biochemical analysis of the clathrin coated pits at the membranes of mammalian cells hinted at a new function for Hsp70 Citation[10], Citation[11]. The HSPs were apparently required to fold the proteins involved in λ page replication and to disassemble the proteins involved in the huge lattice structures of the clathrin coats with the aid of ATP hydrolysis. Hsp70 was described as an ‘unfolding ATPase’, a protein that employs the energy derived from ATP and an intrinsic ATPase activity, to influence quaternary interactions between proteins Citation[11]. HSPs thus became known as ‘molecular chaperones’ due to their role in associating with other proteins and among other things, discouraging promiscuous interactions, and this name has been retained to the present time Citation[10].
During this period (1980–1990) huge strides were also made in elucidating the processes underlying transcriptional regulation of HSP genes. In eukaryotes, a cis-acting element (the heat shock element, or HSE) that confers heat shock regulation on genes was discovered first in Drosophila and then strikingly similar sequences were found in yeast, avian and mammalian cells Citation[12]. Discovery of the HSE sequence was then instrumental in the isolation of the transcription factors (heat shock factor, or HSF) that respond to heat shock, bind to the HSE and activate HSP gene transcription Citation[13]. An excellent review describes the early studies on the refinement of the canonical HSE sequence and the early studies on HSF regulation Citation[14].
In recent years HSPs have been assigned a more empowered role than as chaperones and are now envisaged as central regulators of cell metabolism. This new concept was heralded by the finding that Hsp90 is required for glucocorticoid receptor activity and that, in the absence of Hsp90, GR fails to mature to a transcriptionally active form Citation[15]. It has since been shown that Hsp90 carries out these functions in combination with Hsp70 as well as a host of co-factors (co-chaperones), in the folding of over 100 molecules most of which are signal transduction molecules that must be maintained in a form poised for activation by extracellular or intracellular signals Citation[16]. As an alternative, particularly during heat shock, molecular chaperones can also mark their substrates for a more destructive fate. Hsp70 and Hsp90 bind the ubiquitin E3 ligase CHIP and thus their associated client proteins are tagged with a polyubiquitin chain and delivered to the proteasome for destruction Citation[17]. Heat shock proteins thus function at the cross roads between protein function and destruction.
A long road has thus been traveled since the Drosophila heat shock genes were stumbled upon, leading to the current status of HSP as key physiological intermediates with roles in protein folding and cell regulation in all cellular organisms. We aim here to give an overview of the properties of HSP as molecular chaperones, cell regulators and immunogens that impact on tumor growth and treatment.
Heat shock proteins mediate resistance to hyperthermia
The discovery of a powerful mechanism for hyperthermia resistance (thermotolerance) coincided with the discovery of HSPs and it was soon shown that the onset of thermotolerance coincides quite closely with HSP synthesis Citation[18]. A causal relationship was later shown Citation[19–21]. Thermotolerance is a potent and long-lived form of heat resistance and, for instance a relatively minor pre-exposure to heat such as 15 min at 43°C can lead to profound loss of heat sensitivity Citation[18]. This has had a major impact on how hyperthermia is scheduled when used in combination with radiotherapy, and heating doses have typically been given on a weekly basis to avoid complications due to thermotolerance Citation[22]. The discovery that the HSPs have molecular chaperone activity led to the hypothesis that thermotolerance involves the increased ability to refold heat denatured proteins in cells with elevated HSP levels Citation[23], Citation[24]. Intracellular HSPs were envisaged as limiting protein unfolding during hyperthermia and refolding protein aggregates during the recovery from heat shock. There is a considerable body of evidence in favor of this hypothesis, which considers that a subset of thermally sensitive proteins may confer temperature sensitivity on the cell. Different HSPs appear to chaperone different subsets of proteins Citation[25], Citation[26]. Most of these thermotolerance studies have utilized the clonogenic cell survival assay to quantitate tumor cell inactivation, as this is the most therapeutically relevant parameter.
Heat shock proteins inhibit programmed cell death
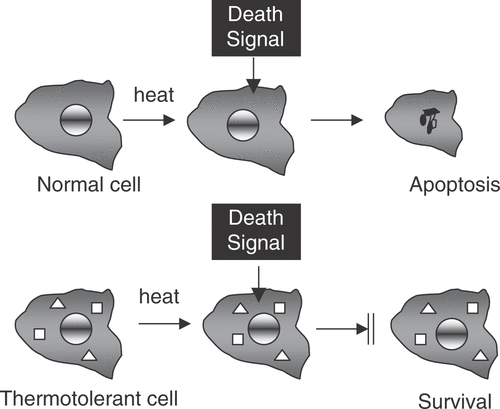
With the increasing understanding of molecular pathways of cell death that began in the later 1980s, it rapidly became evident that HSPs can act directly to inhibit cell death pathways. Hsp70 was shown to inhibit death induced by the stress kinase c-jun kinase Citation[27], Citation[28]. It has recently been shown that Hsp27 and Hsp70 can both inhibit caspase dependent apoptosis pathways. Elevated levels of these HSPs interact directly with intermediates in the death pathways and prevent progression through to the ‘execution stage’ of the cellular death sentence. A number of independent death pathways exist and the HSP appear to act on these through contrasting mechanisms. For instance, Hsp70 inhibits a caspase-independent form of death by interacting with the lysosomal membrane and preventing the activity of hydrolytic lysosomal enzymes Citation[29]. Inhibition of programmed cell death (PCD) by HSP is therefore of key importance in thermotolerance (). Thermotolerance thus involves at least two mechanisms, including: (1) direct inhibition of death pathways and (2) repair of protein damage and resolution of protein aggregates. Inhibition of death may be required to permit the more gradual reactions involved in protein refolding.
Figure 1. Heat shock proteins inhibit programmed cell death. In normal cells stresses such as heat shock induce death signals that trigger programmed cell death. However, when Hsp27 ![]()
The levels of heat shock proteins are elevated in many types of cancer
The levels of Hsp70 and Hsp27 are elevated in a wide spectrum of human cancers and mediate tumorigenesis through mechanisms involving inhibition of PCD, an essential trait in cancer Citation[30–32]. Elevated HSP gene transcription in cancer is coupled to some of the basic oncogenic pathways. A primary mechanism for HSP regulation in normal cells involves the tumor repressor p53 and the related protein p63. These proteins repress HSP transcription through binding sites for the transcription factor NF-Y present within HSP promoters Citation[33], Citation[34]. During transformation, p53 mutation (a genetic change associated with over 45% of cancers at many organ sites) reverses this effect and leads to enhanced Hsp70 transcription through loss of Hsp70 promoter repression Citation[35–38]. Alterations in p63 are closely associated with Hsp70 expression in cancer and expression of the isoform ΔNp63α, a dominant negative inhibitor of wild-type p63 up-regulates Hsp70 and Hsp40 levels in head and neck cancer Citation[33]. In addition to reversal of repression by p53 family proteins, induction of tumor HSP also involves positive effects on transcription through the signaling circuitry of the heat shock response pathway Citation[39]. During the heat shock response massive levels of HSP gene expression occur through interaction of HSF1 with the HSE elements present in all HSP promoters Citation[10], Citation[14], Citation[40]. It was shown recently shown that the tumorigenic factor heregulin-β1 binds to the cell surface of breast cancer cells and leads to increased HSP expression, enhanced survival and transformation through induced stabilization of HSF1 Citation[31]. Heregulin activates HSF1 through a signaling pathway involving activation of HER2 and PI-3 kinase at the cell surface and leads to expression of HSP genes through the HSE elements in their promoters Citation[39]. As PI-3 kinase is a key enzyme in malignant progression, particularly through its activation by PTEN mutation and induction of c-Myc, this may be an important mechanism for HSP elevation in cancer Citation[41]. In addition, the proto-oncogene c-Myc, which is activated by heregulin and HER2 also positively regulates HSP transcription through activation of HSE Citation[34]. Indeed, c-Myc activates the Hsp90A promoter and inhibition of this activation decreases the transforming effects of c-Myc expression Citation[42]. HSF1 also plays a number of additional roles in the malignant phenotype, including the override of cell cycle checkpoints leading to tumor aneuploidy and enhanced metastasis which may involve non-HSP dependent effects of HSF1 Citation[43], Citation[44]. In addition, Hsp90 is the intrinsic repressor of HSF1 under non-stress conditions and one can envisage a mechanism for HSP elevation that includes increased sequestration of Hsp90 by unstable mutated tumor proteins, de-repression of HSF1 resulting in expression of HSP Citation[45]. In addition, Hsp27 transcription is activated by factors in addition to HSF1 including the POU domain protein Brn3a Citation[46–48]. Overall therefore, elevated expression of HSP occurs through relief of repression by p53, which is inactivated in many cancers, and through positive regulation by oncogenic signaling pathways that lead to activation of HSP promoters.
It also seems possible that HSP expression could be induced by the micro-environmental stress imposed by the tumor milieu Citation[49]. However, little information is available to encourage this suggestion and the available data indicate that another transcriptional stress response, the unfolded protein response rather than HSP expression, is activated by the tumor milieu, and growth of tumor cells as xenografts leads to the inhibition rather than enhanced expression of HSP in cells growing in tumors Citation[50].
The levels of Hsp70 and Hsp27 are elevated in a wide spectrum of human cancers and mediate tumorigenesis through involving inhibition of PCD (described above), an essential trait in cancer progression Citation[52–54]. The enhanced activity of a number of oncogenes, most notably c-Myc or Ras, induce PCD pathways including apoptosis and autophagy, and therefore a number of systems involved in PCD regulation including the p53 and Bcl-2 family mediated networks are subverted, and inactivation of these networks are important steps in the emergence of cancer cells Citation[55], Citation[56]. The relationship of Hsp27 and Hsp70 to the p53 and Bcl-2 pathways is currently not clear, although each can function independently in countering death signals Citation[51], Citation[57]. It has been shown that the blockage of cell death through apoptosis and autophagy can lead to a proportion of such cells dying through default by necrosis Citation[58]. This form of death may be less efficient than other death pathways, permitting enhanced growth Citation[56]. In addition, necrosis arising from inhibition of PCD and ischemia in the poorly perfused tumor core is not opposed by Hsp70 overexpression and leads to the release of cell contents into the tumor milieu and initiation of an inflammatory environment in the vicinity of the tumor that favors angiogenesis, tumor cell invasion and metastasis Citation[56], Citation[58], Citation[59].
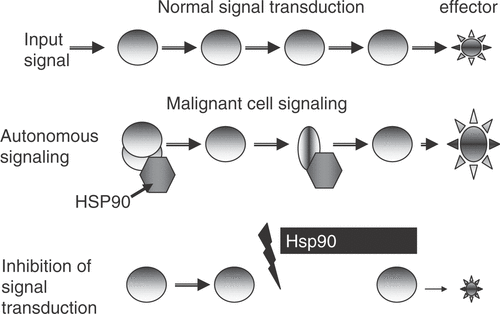
The effects of HSP on the anabolic pathways leading to self-sufficiency in growth signals are mediated largely through Hsp90. This molecular chaperone is essential for the stability of the fragile structures of many of the receptors, protein kinases and transcription factors that lie along the pathways of normal cellular growth Citation[61]. Hsp90 is required to maintain signaling proteins in active conformation that can be rapidly triggered by growth signals Citation[16], Citation[61]. This chaperone may thus be viewed as a facilitator of the rapid and fluid responses to extracellular signals required, particularly in development and cell renewal Citation[61], Citation[62]. Transformation involves the overexpression or mutation of many of these Hsp90-dependent signaling molecules and Hsp90 is increasingly required to maintain such proteins in active conformation. Hsp90 is essential for the stability and activity of over 200 Hsp90 clients Citation[61]. Hsp90 performs these molecular chaperone functions as the dominant component of a high Mr chaperone machine, a large complex incorporating five core proteins found in all complexes: HSP90 itself, the scaffold protein Hop, the p23 protein which mediates substrate choice and a Hsp70/Hsp40 complex that mediates formation of the Hsp90-substrate complexes Citation[62]. Hsp90 complexes are, however, heterogeneous and steroid hormone receptors, for instance, contain in addition to the core proteins the immunophilins FKBP51, FKBP52 and CyP40 necessary for receptor function Citation[62]. Pharmacological targeting of Hsp90 using specific chemical inhibitors leads to the degradation of the client proteins and inhibition of tumor growth through G1 arrest, morphological and functional differentiation and activation of apoptosis Citation[61] (). This strongly implicates Hsp90 as a key component required for ‘self sufficiency in growth signals’. However, overexpression of Hsp90 in tumor cells compared to normal tissues has been observed only in sporadic cases Citation[30], Citation[42]. More subtle alterations are also involved. For example, the splice variant Hsp90N which lacks an ATP binding site is observed in some cancers and mediates transformation Citation[63], Citation[64]. In addition, the co-chaperone cdc37 which is essential for the function of a subset of growth-related, Hsp90 binding protein kinases in normal cells is an oncogene in itself when overexpressed in prostate carcinoma Citation[65]. Cdc37 binding to protein kinases and cyclophilin binding to nuclear receptors are mutually exclusive interactions, pointing to the existence of unique classes of individual Hsp90 complexes that could be targeted in therapy. Changes in the abundance and composition of Hsp90 complexes in cancer increase the chaperoning reservoir needed to foster oncogenic proteins Citation[66–69]. In addition, the increased susceptibility of Hsp90 in tumors to ansamycin family drugs (which target their ATPase domains) reflects the concentration of tumor Hsp90 within the chaperone machine complexes in which form it has a high affinity for the drugs as opposed to ‘free Hsp90’ in unbound form which predominates in normal cells and has low affinity for drugs Citation[69].
Figure 2. Hsp90 inhibitors can preferentially reduce growth signaling in malignant cells. In normal cells, growth requires input from receptors coupled to extracellular signaling molecules. Input signals are amplified by coupled signaling enzymes ![]()
Extracellular HSP; carriers of tumor antigens and vaccine components
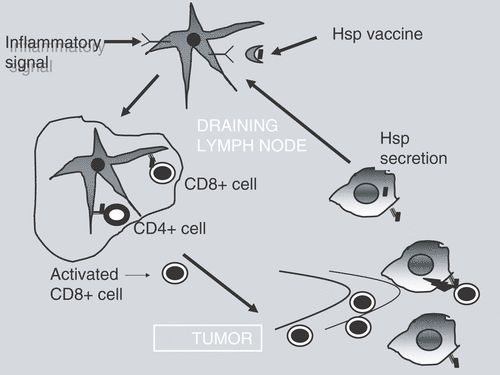
Heat shock proteins have recently been shown to play important roles in the immune system as carriers of tumor antigens and inflammatory agents Citation[70], Citation[71] (). Such HSPs are able to form complexes with peptide antigens in the cytoplasm, which can then be secreted and may ultimately participate in immune surveillance. Extracellular Hsp70 interacts with receptors on antigen presenting cells (APC) either during episodes of cell death and lysis in vivo or after treatment with molecular chaperone-based vaccines. Hsp70-peptide complexes are thus able to deliver antigens to major histocompatibility (MHC) class I and II molecules on the APC cell surface and present such tumor antigens to T lymphocytes. HSP-antigen complexes have proven effective in the treatment of rodent tumors in preclinical studies and are now undergoing clinical trial for treatment of human cancer.
Figure 3. Heat shock proteins induce specific immune killing of tumor cells. HSP ![]()
The pioneer in molecular chaperone-based anticancer vaccine design has been Pramod Srivastava who has prepared autologous vaccines in mice and in human patients with the aim of directly targeting the unique, often mutated antigens that characterize each individual neoplasm Citation[72–76]. In this approach, HSP.PC (Hsp70 and gp96) are isolated from the patients’ tumors by affinity chromatography and formulations of such purified HSP are then applied in a multi-dose regimen. The aim is for tumor antigens to be cross-presented to the patient's APC and for the unique mixture of peptides from the individual tumor to induce immunity. These studies are currently undergoing Phase III trial and recent results suggest a trend towards increased response particularly in patients receiving longer courses of vaccination Citation[77]. Success may thus be limited by the amount of tumor available after resection as response to treatment evidently is related to the number of treatments with HSP vaccine Citation[77]. These results may be viewed as encouraging when compared with the recent NCI trials of metastatic cancer treatment in the United States with synthetic peptides, DNA vaccines, dendritic cell vaccines and viruses in which an objective response rate of 2.6% was reported Citation[78]. Factors that limit the effects of vaccines may be structural and relate to the avidity with which individual HSPs bind to peptides, the nature of their peptide repertoires and ability to induce a co-stimulatory response. For instance Hsp70 gene family members Hsp110 and grp170 possess a greatly enhanced ability to bind peptides compared with other HSPs, can bind avidly to larger polypeptides, and are superior agents in cancer vaccine production than smaller HSPs Citation[79], Citation[80]. Recent studies suggest that larger polypeptides are superior to smaller peptides in inducing immunity Citation[81], Citation[82].
Another recent approach emphasizes use of combinations of chaperones with the hope of increasing the repertoire of peptides presented to APC. Indeed the studies of Binder and Srivastava (2005) indicate that Hsp70, gp96, Hsp90 and calreticulin carry between them the whole of the intracellular peptide repertoire required for cross priming T cells against ovalbumin or β-galactosidase. An approach using chaperone-rich lysates has been described in which tumor cell lysates are partially purified by iso-electric focusing to enrich the above chaperones Citation[83]. Such preparations are effective against mouse tumors in prophylactic context. These studies are supported by a recent publication showing that the effectiveness of an Hsp70-based vaccine derived from tumor-DC fusion cells is partially dependent on co-isolation of Hsp90 Citation[84]. Use of multi-chaperone formulations may thus be indicated. These studies used MUC1, a normal antigen whose expression and processing is altered in cancer. Mice expressing MUC1 became tolerant to this antigen while the Hsp70-based vaccine was able to overcome tolerance and cause tumor rejection, suggesting that non-mutated antigens may be targeted by Hsp70-based vaccines Citation[84].
It has been suggested that there are three requirements for effective cancer immunotherapy: (1) a sufficient number of avid tumor reactive lymphocytes present in the tumor bearing host, (2) these must be capable of reaching the tumor and extravasating, (3) lymphocytes that penetrate the tumor must have appropriate effector mechanisms to destroy the cancer Citation[85]. One innovative approach described recently emphasizes the inflammatory nature of HSP; local inflammation may lead to increased influx of T lymphocytes and enhanced cell killing due to APC maturation. The approach devised by Vile et al. involves targeted killing of normal melanocytes overexpressing HSP70 to generate an antigen-specific CD8+ T lymphocyte response against established melanoma Citation[86], Citation[87]. The rationale behind this unorthodox approach is that the majority of peptides that are presented by melanoma cells and recognized by T cells from patients arise from developmental proteins that are also expressed in normal melanocytes Citation[88]. The existence of shared antigens between the proliferating melanocytes and the melanoma cells suggested that if a CD8+ T cell response could be generated against the dying melanocytes it could also target the tumor cells Citation[86]. This approach requires the specific targeting of proliferating melanocytes using local expression of a cell suicide gene (HSVtk) under the control of a promoter from the tyrosinase gene that is specifically active in this cell population. For tumor rejection, the critical requirement was that killing take place in melanocytes engineered to overexpress Hsp70 Citation[86]. Cell death in the sacrificed population leads to the extracellular release of the Hsp70, modulation of DC function, and generation of a CD8+ T cell response against melanocytic TSTA that eradicates primary and metastatic melanoma Citation[86]. The preclinical studies in mice show the feasibility of this approach and its potential for translation for clinical treatment of malignant melanoma Citation[86]. One further question is whether such an approach could be generalized to the treatment of other tumors at different sites. This approach would require the targeted sacrifice of a population of normal cells with an antigenic repertoire similar to the tumor under treatment. A related approach involves local killing of tumor cells with forced overexpression of Hsp70, obviating the need to find a related normal tissue for priming antitumor immunity Citation[89], Citation[90]. Of course, the question of potential autoimmune destruction of normal tissues, especially as this approach depends on the generation of CD8+ T lymphocytes directed against antigens common to normal cells and tumor cells is a concern with this approach Citation[86]. The targeting of the small fraction of proliferating melanocytes may, however, limit the extent of normal cell targets. In addition, the studies of Vile et al. show that the effects of the treatment are self-regulatory, through the activation of CD4+CD25+ lymphocytes, which inhibited the activity of tumor-specific CD8+ T lymphocytes and rapidly attenuated the response Citation[86].
A further refinement is to combine chaperone-based immunotherapy with low temperature hyperthermia Citation[91]. Fever-range hyperthermia has the advantage of causing HSP induction and release and inducing highly efficient homing of APC and T lymphocytes to tumors and activation Citation[92], Citation[93]. Recent studies show that hyperthermia also causes lymphocyte trafficking across the high endothelial venules through a mechanism involving enhanced IL-6 signaling Citation[94]. This approach therefore is of high promise in combination with HSP-based immunotherapy. Finally, in another novel approach, HSPs and peptide/protein antigens derived from the tumor have been combined with ceramic microparticles to produce an autologous anticancer vaccine. In this case, the ceramic particles were used to purify several HSPs and when injected into mice attracted APC. This therapeutic vaccine is showing encouraging clinical results Citation[95].
Conclusions
Heat shock proteins are stress proteins with Janus-like properties in cancer
The HSPs play a fairly unambiguous role in mediating the response to stress. Elevated levels of HSP lead to resistance in almost all contexts. However, their role in cancer is less straightforward. Elevated HSP levels may be required in the early stages of cancer to counter the induction of programmed cell death accompanying transformation. There may thus be selection for cells expressing high levels of HSP. Such cells would be expected to be resistant to therapies that function through targeting programmed cell death. In more advanced cancers. There may, however, be immune selection for cells with lower HSP levels in order to escape immune surveillance Citation[96]. More information is required in order to resolve these questions. This dichotomy is also reflected in approaches to targeting HSP in therapy. Pharmacological approaches aim to inhibit activity or expression of tumor HSP and in the case of Hsp90 this is a highly effective approach. Immunological approaches aim at increasing HSP levels in cells with the aim of increasing tumor antigen presentation to the immune system. These contrasting approaches to therapy therefore mirror the Janus-like properties of the HSP in the development of cancer.
References
- Ellis RJ. Molecular chaperones: Assisting assembly in addition to folding. Trends Biochem Sci 2006; 31: 395–401
- Ritossa F. A new puffing pattern induced by temperature shock and DNP in Drosophila. Experientia 1962; 18: 571–573
- Ashburner M. The effects of heat shock and other stresses on gene activity: An introduction. Cold Spring Harbor laboratory Publications, Cold Spring Harbor Laboratory 1982
- Barnett TM, Altschuler CN, McDaniel CN, et al. Heat shock induced proteins in plant cells. Dev Genet 1980; 1: 331–340
- Bouche G, Amalric F, Caizergues-Ferrer M, et al. Effects of heat shock on gene expression and subcellular protein distribution in Chinese hamster ovary cells. Nucleic Acids Res 1979; 7: 1739–1747
- Kelley PM, Schlesinger MJ. The effect of amino acid analogues and heat shock on gene expression in chicken embryo fibroblasts. Cell 1978; 15: 1277–1286
- Lemeaux PG, Herendeen SL, Bloch PL, et al. Transient rates of synthesis of individual polypeptides in E. coli following temperature shifts. Cell 1978; 13: 427–434
- Miller MJ, Xuong N-H, Geiduschek EP. A response of protein synthesis to temperature shift in the yeast Saccharomyces cerevisiae. Proc Nat Acad Sci USA 1979; 76: 1117–1121
- Hightower LE, White FP. Cellular responses to stress: comparison of a family of 71–73-kilodalton proteins rapidly synthesized in rat tissue slices and canavanine-treated cells in culture. J Cell Physiol 1981; 108: 261–275
- Georgopolis C, Welch WJ. Role of the major heat shock proteins as molecular chaperones. Ann Rev Cell Biol 1993; 9: 601–634
- Schlossman DM, Schmid SL, Braell WA, et al. An enzyme that removes clathrin coats: Purification of an uncoating ATPase. J Cell Biol 1984; 99: 723–733
- Pelham HR, Bienz M. A synthetic heat-shock promoter element confers heat-inducibility on the herpes simplex virus thymidine kinase gene. Embo J 1982; 1: 1473–1477
- Sorger PK, Pelham HR. Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell 1988; 54: 855–864
- Wu C. Heat shock transcription factors: Structure and regulation. Ann Rev Cell Dev Biol 1995; 11: 441–469
- Picard D, Khursheed B, Garabedian MJ, et al. Reduced levels of Hsp90 compromise steroid receptor action in vivo. Nature 1990; 348: 166–168
- Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the Hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003; 228: 111–133
- Connell P, Ballinger CA, Jiang J, et al. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol 2001; 3: 93–96
- Li GC, Hahn GM. A proposed operational model for thermotolerance. Cancer Research 1981; 40: 4501–4508
- Subjeck JR, Sciandra JJ, Johnson RJ. Heat shock proteins and thermotolerance; a comparison of induction kinetics. Br J Radiol 1982; 55: 579–584
- Li GC, Werb Z. Correlation between the synthesis of heat shock proteins and the development of thermotolerance in CHO fibroblasts. Proc Nat Academy of Sci USA 1982; 79: 3218–3222
- Landry J, Bernier D, Chretien P, et al. Synthesis and degradation of heat shock proteins during the development and decay of thermotolerance. Cancer Research 1982; 42: 2457–2461
- Sapareto S, Dewey WC. Thermal dose determination in cancer therapy. Int J Radiat Oncol Biol Phys 1984; 10: 787–800
- Kampinga HH, Brunsting JF, Stege GJ, et al. Thermal protein denaturation and protein aggregation in cells made thermotolerant by various chemicals: Role of heat shock proteins. Exp Cell Res 1995; 219: 536–546
- Kampinga HH, Brunsting JF, Stege GJ, et al. Cells overexpressing Hsp27 show accelerated recovery from heat-induced nuclear protein aggregation. Biochem Biophys Res Commun 1994; 204: 1170–1177
- Buchner J. Hsp90 & Co. - A holding for folding. TIBS 1999; 24: 136–142
- Wegele H, Muller L, Buchner J. Hsp70 and Hsp90 - A relay team for protein folding. Rev Physiol Biochem Pharmacol 2004; 151: 1–44
- Gabai VL, Meriin AB, Mosser DD, et al. Hsp70 prevents activation of stress kinases. A novel pathway of cellular thermotolerance. J Biol Chem 1997; 272: 18033–18037
- Gabai VL, Meriin AB, Yaglom JA, et al. Role of Hsp70 in regulation of stress-kinase JNK: Implications in apoptosis and aging. FEBS Lett 1998; 438: 1–4
- Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett 2007; 581: 3702–3710
- Ciocca DR, Calderwood SK. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment applications. Cell Stress Chaperones 2005; 10: 86–103
- Calderwood SK, Khaleque MA, Ciocca DR. Heat shock proteins in the progression of cancer. Springer, New York 2007; 7
- Calderwood SK, Khaleque MA, Sawyer DB, et al. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem Sci 2006; 31: 164–172
- Wu G, Osada M, Guo Z, et al. DeltaNp63alpha up-regulates the Hsp70 gene in human cancer. Cancer Res 2005; 65: 758–766
- Taira T, Sawai M, Ikeda M, et al. Cell cycle-dependent switch of up- and down-regulation of human Hsp70 gene expression by interaction between c-Myc and CBF/NF-Y. J Biol Chem 1999; 274: 24270–24279
- Tsutsumi-Ishii Y, Tadokoro K, Hanaoka F, et al. Response of heat shock element within the human Hsp70 promoter to mutated p53 genes. Cell Growth Diff 1995; 6: 1–8
- Madden SL, Galella EA, Zhu J, et al. SAGE transcript profiles for p53-dependent growth regulation. Oncogene 1997; 15: 1079–1085
- Ghioni P, Bolognese F, Duijf PH, et al. Complex transcriptional effects of p63 isoforms: Identification of novel activation and repression domains. Mol Cell Biol 2002; 22: 8659–8668
- Agoff SN, Hou J, Linzer DI, et al. Regulation of the human Hsp70 promoter by p53. Science 1993; 259: 84–87
- Khaleque MA, Bharti A, Sawyer D, et al. Induction of heat shock proteins by heregulin beta1 leads to protection from apoptosis and anchorage-independent growth. Oncogene 2005; 24: 6564–6573
- Lindquist S, Craig EA. The heat shock proteins. Ann Rev Genet 1988; 22: 631–637
- Bader AG, Kang S, Zhao L, et al. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer 2005; 5: 921–929
- Teng SC, Chen YY, Su YN, et al. Direct activation of Hsp90A transcription by c-Myc contributes to c-Myc-induced transformation. J Biol Chem 2004; 279: 14649–14655
- Wang Y, Theriault JR, He H, et al. Expression of a dominant negative heat shock factor-1 construct inhibits aneuploidy in prostate carcinoma cells. J Biol Chem 2004; 279: 32651–32659
- Hoang AT, Huang J, Rudra-Ganguly N, et al. A novel association between the human heat shock transcription factor 1 (HSF1) and prostate adenocarcinoma. Am J Pathol 2000; 156: 857–864
- Zou J, Guo Y, Guettouche T, et al. Repression of heat shock transcription factor HSF1 activation by Hsp90 (Hsp90 complex) that forms a stress-sensitive complex with HSF1. Cell 1998; 94: 471–480
- Arrigo AP. Heat shock proteins as molecular chaperones. 2005; 21: 619–625, Med Sci (Paris)
- Farooqui-Kabir SR, Budhram-Mahadeo V, Lewis H, et al. Regulation of Hsp27 expression and cell survival by the POU transcription factor Brn3a. Cell Death Differ 2004; 11: 1242–1244
- Lee SA, Ndisang D, Patel C, et al. Expression of the Brn-3b transcription factor correlates with expression of Hsp27 in breast cancer biopsies and is required for maximal activation of the Hsp27 promoter. Cancer Res 2005; 65: 3072–3080
- Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol 2002; 29: 15–18
- Tang D, Khaleque AA, Jones ER, et al. Expression of heat shock proteins and HSP messenger ribonucleic acid in human prostate carcinoma in vitro and in tumors in vivo. Cell Stress Chaperones 2005; 10: 46–59
- Beere HM. Stressed to death: Regulation of apoptotic signaling pathways by the heat shock proteins. Sci STKE 2001, 2001:RE1
- Nylandsted J, Brand K, Jaattela M. Heat shock protein 70 is required for the survival of cancer cells. Ann NY Acad Sci 2000; 926: 122–125
- Tenniswood MP, Guenette RS, Lakins J, et al. Active cell death in hormone-dependent tissues. Cancer Metastasis Rev 1992; 11: 197–220
- Paul C, Manero F, Gonin S, et al. Hsp27 as a negative regulator of cytochrome C release. Mol Cell Biol 2002; 22: 816–834
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70
- Nelson DA, White E. Exploiting different ways to die. Genes Dev 2004; 18: 1223–1226
- Jaattela M. Multiple cell death pathways as regulators of tumour initiation and progression. Oncogene 2004; 23: 2746–2756
- Proskuryakov SY, Konoplyannikov AG, Gabai VL. Necrosis: A specific form of programmed cell death?. Exp Cell Res 2003; 283: 1–16
- Viatour P, Merville MP, Bours V, et al. Phosphorylation of NF-kappaB and IkappaB proteins: Implications in cancer and inflammation. Trends Biochem Sci 2005; 30: 43–52
- Ciocca DR, Oesterreich S, Chamness GC, et al. Biological and clinical implications of heat shock protein 27,000 (Hsp27): A review. J Natl Cancer Inst 1993; 85: 1558–1570
- Neckers L, Ivy SP. Heat shock protein 90. Curr Opin Oncol 2003; 15: 419–424
- Pratt WB, Galigniana MD, Harrell JM, et al. Role of Hsp90 and the Hsp90-binding immunophilins in signalling protein movement. Cell Signal 2004; 16: 857–872
- Zhou V, Han S, Brinker A, et al. A time-resolved fluorescence resonance energy transfer-based HTS assay and a surface plasmon resonance-based binding assay for heat shock protein 90 inhibitors. Anal Biochem 2004; 331: 349–357
- Grammatikakis N, Vultur A, Ramana CV, et al. The role of Hsp90N, a new member of the Hsp90 family, in signal transduction and neoplastic transformation. J Biol Chem 2002; 277: 8312–8320
- Pearl LH. Hsp90 and Cdc37–A chaperone cancer conspiracy. Curr Opin Genet Dev 2005; 15: 55–61
- Scheibel T, Buchner J. The Hsp90 complex–A super-chaperone machine as a novel drug target. Biochem Pharmacol 1998; 56: 675–682
- Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med 2002; 8: S55–61
- Neckers L, Lee YS. Cancer: The rules of attraction. Nature 2003; 425: 357–359
- Kamal A, Thao L, Sensintaffar J, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003; 425: 407–410
- Calderwood SK, Theriault JR, Gong J. Message in a bottle: Role of the 70-kDa heat shock protein family in anti-tumor immunity. Eur J Immunol 2005; 35: 2518–2527
- Calderwood SK, Theriault JR, Gong J. How is the immune response affected by hyperthermia and heat shock proteins?. Int J Hyperthermia 2005; 21: 713–716
- Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: Chaperoning of the innate and adaptive immune responses. Annu Rev Immunol 2002; 20: 395–425
- Srivastava P. Hypothesis: Controlled necrosis as a tool for immunotherapy of human cancer. Cancer Immun 2003; 3: 4
- Srivastava PK. Immunotherapy of human cancer: Lessons from mice. Nat Immunol 2000; 1: 363–366
- Belli F, Testori A, Rivoltini L, et al. Vaccination of metastatic melanoma patients with autologous tumor-derived heat shock protein gp96-peptide complexes: Clinical and immunologic findings. J Clin Oncol 2002; 20: 4169–4180
- Mazzaferro V, Coppa J, Carrabba MG, et al. Vaccination with autologous tumor-derived heat-shock protein gp96 after liver resection for metastatic colorectal cancer. Clin Cancer Res 2003; 9: 3235–3245
- Srivastava PK. Therapeutic cancer vaccines. Curr Opin Immunol 2006; 18: 201–205
- Rosenberg SA, Yang JC, Restifo NP. Cancer Immunotherapy: Moving beyond current vaccines. Nature Medicine 2004; 10: 909–915
- Manjili MH, Wang XY, Chen X, et al. Hsp110-HER2/neu chaperone complex vaccine induces protective immunity against spontaneous mammary tumors in HER-2/neu transgenic mice. J Immunol 2003; 171: 4054–4061
- Segal BH, Wang XY, Dennis CG, et al. Heat shock proteins as vaccine adjuvants in infections and cancer. Drug Discov Today 2006; 11: 534–540
- Rock KL, Hearn A, Chen CJ, et al. Natural endogenous adjuvants. Springer Semin Immunopathol 2005; 26: 231–246
- Shen L, Rock KL. Priming of T cells by exogenous antigen cross-presented on MHC class I molecules. Curr Opin Immunol 2006; 18: 85–91
- Zeng Y, Graner MW, Katsanis E. Chaperone-rich cell lysates, immune activation and tumor vaccination. Cancer Immunol Immunother 2006; 55: 329–338
- Enomoto Y, Bharti A, Khaleque A, et al. Enhanced immunogenicity of Hsp70 peptide complexes from DC-tumor fusion cells requires Myd-88. J Immunol 2006, (in press)
- Rosenberg SA. Shedding light on tumor immunotherapy of cancer. NEJM 2004; 350: 1461–1463
- Daniels GA, Sanchez-Perez L, Diaz RM, et al. A simple method to cure established tumors by inflammatory killing of normal cells. Nat Biotechnol 2004; 22: 1125–1132
- Calderwood SK. Chaperones and slow death–A recipe for tumor immunotherapy. Trends Biotechnol 2005; 23: 57–59
- Engelhard VH, Bullock TN, Colella TA, et al. Antigens derived from melanocyte differentiation proteins: Self-tolerance, autoimmunity, and use for cancer immunotherapy. Immunol Rev 2002; 188: 136–146
- Todryk S, Melcher AA, Hardwick N, et al. Heat shock protein 70 induced during tumor cell killing induces Th1 cytokines and targets immature dendritic cell precursors to enhance antigen uptake. J Immunol 1999; 163: 1398–1408
- Huang XF, Ren W, Rollins L, et al. A broadly applicable, personalized heat shock protein-mediated oncolytic tumor vaccine. Cancer Res 2003; 63: 7321–7329
- Repasky E, Issels R. Physiological consequences of hyperthermia: Heat, heat shock proteins and the immune response. Int J Hyperthermia 2002; 18: 486–489
- Mambula SS, Calderwood SK. Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J Immunol 2006; 177: 7849–7857
- Evans SS, Wang WC, Bain MD, et al. Fever-range hyperthermia dynamically regulates lymphocyte delivery to high endothelial venules. Blood 2001; 97: 2727–2733
- Chen Q, Fisher DT, Clancy KA, et al. Fever-range thermal stress promotes lymphocyte trafficking across high endothelial venules via an interleukin 6 trans-signaling mechanism. Nat Immunol 2006; 12: 1299–1308
- Ciocca DR, Frayssinet P, Cuello-Carrión FD. A pilot study with a therapeutic vaccine based on hydroxyapatite ceramic particles and self-antigens in cancer patients. Cell Stress Chaperones 2007; 12: 33–43
- Bonorino C, Souza AP. Hsp70 in tumors: Friend or foe?. Heat shock proteins in biology and medicine, SK Calderwood, MY Sherman, DR Ciocca. Springer, New York 2007