
Abstract
Purpose: Modulated electro-hyperthermia (mEHT) has been shown to be effective against various types of human tumours, including hepatocellular carcinoma (HCC). Here we aimed to investigate the molecular mechanism underlying the cytotoxic effects of mEHT to HCC cells.
Materials and methods: Human liver cancer cell lines, Huh7 and HepG2, were treated with mEHT (42 °C/60 min) three times at 2-day intervals. Growth inhibition and apoptotic induction were evaluated using MTS, microscopic analysis, a clonogenic assay, annexin V/PI staining and a ccK18 ELISA. Global changes in gene expression were examined using RNA sequencing to obtain insights into molecular changes in response to mEHT. For in vivo evaluation of mEHT we used HepG2 HCC xenografts grown in nude mice.
Results: mEHT suppressed HCC cell proliferation and long-term colony formation through induction of apoptosis. The growth inhibitory effects are induced through a subset of molecular changes. Notably the expression level of septin 4 (SEPT4) (involved in pro-apoptotic activity and growth suppression) was up-regulated, whereas a key regulator of invasiveness G-Protein coupled receptor 64 (GPR64) was repressed. Subsequent Western blotting confirmed that the common increase in tumour suppressor SEPT4 in both Huh7 and HepG2 cells is accompanied by the restoration of cyclin-dependent kinase (CDK) inhibitor p21 and decrease in pro-caspase 7 and pro-caspase 3, thereby accelerating apoptotic signalling in HCC cells. Additionally, mEHT significantly inhibited the growth of human HCC xenografts in nude mice.
Conclusions: These findings suggest that apoptotic cell death induced by mEHT is mediated by the up-regulation of tumour suppressor SEPT4 in human HCC cells.
Keywords:
Introduction
HCC is the sixth most commonly diagnosed and the third most lethal neoplasm and causes an estimated 700,000 deaths annually worldwide [Citation1]. The incidence of HCC has doubled over the past two decades, especially in the USA, Europe, Japan and China. Similarly, about 14,000 annual incidences of HCC have been reported in South Korea. HCC is predominantly associated with risk factors with geographic variations, including HBV and HCV infections, alcohol abuse, obesity and type 2 diabetes [Citation2]. According to the Barcelona Clinic Liver Cancer (BCLC) staging system, curative treatments such as surgical resection, liver transplantation or RFA-PEI treatment are primarily suitable for asymptomatic patients with very early and early (both stage A) HCCs; these treatments extend the median overall-survival (OS) by more than 60 months [Citation2,Citation3]. The DC-BEAD transarterial chemoembolisation (TACE) method is primarily curative for patients in the intermediate and multinodular stage B, with a median OS of approximately 20 months. Sorafenib is the only recommended treatment for patients with advanced HCC (stage C) with invasive or extrahepatic tumours. However, the standard-of-care for advanced liver cancer can extend the median OS for only 12 months. Overall, only 30 ∼ 40% of HCC patients are diagnosed as eligible for current curative treatments due to late diagnosis, underlying liver disease and a lack of effective treatment options [Citation4].
Hyperthermia cancer therapy is presently used either for ablation purposes as an alternative to surgery or in combination with chemotherapy and/or radiation therapy to augment the effects of those traditional therapies [Citation5,Citation6]. The principle of hyperthermia is to achieve tumour temperatures in the range of 40-46 °C by controlling temperature alone [Citation7]. More recently developed mEHT is based on the principle of classical hyperthermia, except that mEHT is physically designed to give an energy dose by regulating radiofrequency (RF) power level with an electronic device, replacing the single temperature concept of hyperthermia [Citation7–10]. mEHT promotes direct absorption of the modulated RF current into the extracellular fluid, creating a slight temperature difference (1/1000 °C) between the inner and outer temperature of the cell. Therefore the heat flows into the cell through the membrane until a temperature difference disappears, thereby destroying the membrane. In principle, the enriched electronic field and heat flow can selectively induce cell death in cancer cells with elevated glycolysis, higher level of lactate production due to Warburg effect and reduced electronic impedance when compared to normal cells.
mEHT in combination with the molecular targeted agent sorafenib exhibited synergistic anti-cancer effects in clinical trials with hepatocellular carcinoma patients [Citation11]. In clinical settings the anti-HCC efficacy of mEHT has been studied, but the underlying molecular mechanisms of this treatment for HCC are not yet clearly understood. In this study our goal was to elucidate the molecular mechanisms mediating the phenotypic changes in oncothermic HCC cells using a laboratory mEHT device for in vitro experiments and an HCC xenograft model. Further, by using RNA sequencing and subsequent protein analysis, we investigated the mechanism on a whole genome scale. mEHT suppressed HCC cell proliferation and long-term colony formation and induced apoptosis. The growth inhibitory effects were induced through a subset of common molecular alterations including the up-regulation of SEPT4 and CDK inhibitor p21, which play a pivotal role in apoptotic progression. We also observed that mEHT effectively suppressed the growth of human HCC xenografts in nude mice. These results demonstrate that mEHT is an alternative treatment option for HCC and that apoptotic cell death caused by mEHT is mediated by the up-regulation of tumour suppressor SEPT4 in HCC cells.
Materials and methods
Cell culture
The human HCC cell lines Huh7 and HepG2 were purchased from the Japanese Collection of Research Biosources Cell Bank (JCRB, Osaka). Huh7 and HepG2 cells were cultured in DMEM medium (HyClone, South Logan, UT, USA) supplemented with 10% fetal bovine serum (HyClone) and 1% penicillin/streptomycin solution (HyClone) at 37 °C with 5% CO2. The phenotypes of these cell lines have been authenticated by the JCRB.
Modulated electro-hyperthermia (mEHT)
The LAB-EHY100 device (OncoTherm, Budapest, Hungary) was used for in vitro and in vivo mEHT treatment. To mimic clinical conditions in vitro, 1 day prior to initiating mEHT, Huh7 and HepG2, cells were seeded in microscope cover glasses (1.5 × 105 cells per cover glass, Marienfeld, Lauda-Konigshofen, Germany), and the cover glasses were submerged into 60 mm-culture dishes containing culture media and incubated for 24 h in a 5% CO2 incubator. The next day the glasses with the attached HCC cells were loaded into a treatment cup with culture media and exposed to mEHT (3 W power, 42 °C for 60 min). The RF power level was controlled during the 60 min of mEHT, using the fluorotopic temperature measurement system (Luxton m3300; Lumasense, Santa Clara, CA), to maintain the heating temperature at 42 °C. After the 1-h treatment, the cover glasses were placed back in the culture dishes for continuous incubation until the next treatment. We have repeated this treatment three times with a 2-day interval. After the third mEHT treatment the heated cells were detached from the cover glasses by trypsinisation and then re-seeded into a 96-well plate (2000 cells per well) for subsequent cell proliferation assay and microscopic observation. Aliquots of 50 μg of the extract from heated cells were applied to each well of a 96-well plate for the detection of ccK18 fragments by ELISA. Furthermore, after the third mEHT 1000 HCC cells were re-seeded into each well of 6-well plates for a clonogenic assay and 1.0 × 105 cells were directly applied for the detection of apoptotic cells by FACS analysis. To elucidate the efficacy of mEHT against in vivo tumours, 1 × 107 HepG2 cells were injected into the right flanks of 4-week-old male BALB/c nude mice (Orientbio, Seongnam, Korea). The xenografts were treated with mEHT conditions 9, 11, 13, 15, and 17 days after tumour cell injection. The tumour size was measured using a vernier caliper and calculated as the minor axis2 × the major axis ×1/2.
Measurement of cell proliferation and detection of apoptosis
Cell growth was measured 72 h after the third mEHT using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The cells were incubated for 2 h at 37 °C in a humidified 5% CO2 atmosphere. The production of coloured formazan products was determined by measuring the absorbance at 490 nm using a VersaMax microplate reader (Molecular Devices, Sunnyvale, CA). The detection of apoptosis was analysed by FACS at 48 h and 72 h after the third mEHT using the FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen, San Jose, CA) according to the manufacturer’s instructions. The cell death was measured through FACSVerse flow cytometry (BD Bioscience, Franklin Lakes, NJ) and was quantified using FlowJo software (Ashland, OR). The induction of apoptosis was also detected 48 h after the third mEHT by measuring the level of caspase-cleaved keratin 18 (ccK18) fragments using the M30 Apoptosense ELISA Kit (PEVIVA, Stockholm, Sweden) as recommended by the manufacturer.
Clonogenic assay
The effect of mEHT on the clonogenic survival of tumours was studied. Cells treated with mEHT were re-seeded into 6-well culture plates at a density of 1 × 103 cells per well. The cells were maintained in culture for 12 days without a medium change to allow the viable cells to propagate to sizable colonies for quantification. The colonies were fixed with 4% paraformaldehyde and then stained with 0.5% crystal violet for 30 min at room temperature. The number of colonies containing more than 50 cells was counted.
RNA sequencing
Total RNA was extracted 48 h after the third mEHT using the RNeasy Mini Kit (Qiagen, Valencia, CA). The quantity of the total RNA was evaluated using RNA electropherograms (Bio-Rad Experion, Hercules, CA); RNA quality was assessed based on the RNA quality indicator (RQI). The total RNA from each sample with a RQI value of 8.0 or higher was used. The resulting mRNA samples were processed for sequencing libraries using the Illumina TruSeq Stranded mRNA Sample Preparation Kit (Illumina, San Diego, CA) according to the manufacturer’s protocols. RNA sequencing was performed using the Illumina HiSeq 2500 to generate non-directional, paired-end 100-base-pair reads. Quality-filtered reads were mapped to the human reference genome sequence hg19 (UCSC Genome Bioinformatics, https://genome.ucsc.edu) using TopHat2 (http://ccb.jhu.edu/software/tophat). The relative transcript abundance was estimated by counting the fragments per kilobase of the exon model per million mapped sequence reads (FPKM), and differentially expressed genes were evaluated using the cufflinks package (http://cole-trapnell-lab.github.io/cufflinks). The significantly overlapping pathways and gene ontology categories with differentially expressed genes were analysed using DAVID (http://david.abcc.ncifcrf.gov).
Western blot analysis
Cells were suspended in RIPA buffer (Thermo Scientific, Rockford, IL) containing 0.01% of a protease and phosphatase inhibitor cocktail (Thermo Scientific) 24 h after the third mEHT. Equal amounts (80 μg) of total proteins were fractionated by SDS-PAGE on a 10% gel and transferred to polyvinylidene difluoride membranes (Roche, Basel, Switzerland). The membrane was blocked with 5% milk/Tris-buffered saline plus Tween 20 (TBST) and incubated with primary antibodies against human SEPT 4 (sc-20179), p53 (sc-6243), pro-caspase 7 (sc-56063), pro-caspase 3 (sc-7272) (all from Santa Cruz Biotechnology, Santa Cruz, CA), p21 (no. 2947 from Cell signaling, Danvers, MA), and Bax (ab32503, Abcam, Cambridge, UK). HRP goat anti-mouse IgG and HRP goat anti-rabbit IgG (Santa Cruz Biotechnology) were used as the secondary antibodies. Immunoreactive bands were visualised with an LAS-3000 Imager (Fujifilm Corporation, Tokyo, Japan). Equal loading was assessed by probing the same membrane with a β-actin antibody (Santa Cruz Biotechnology). The intensity of protein bands were measured by ImageJ software (US National Institutes of Health, Bethesda, MD). Relative band intensities between the control and the mEHT treatment were then numerically calculated after normalising to β-actin protein expression.
Statistical analysis
Statistical comparisons were conducted using Student’s t-test with equal variance, Wilcoxon rank sum test, bootstrap ANOVA with contrast test with 10,000 random repetitions, or normality assumption test. All data are shown as the means ± SEM. Analyses were conducted using the R statistical software (v. 3.0.1). P values ≤0.05 (*) and ≤0.01 (**) were considered statistically significant.
Results
mEHT inhibits proliferation and induces apoptosis in HCC cells
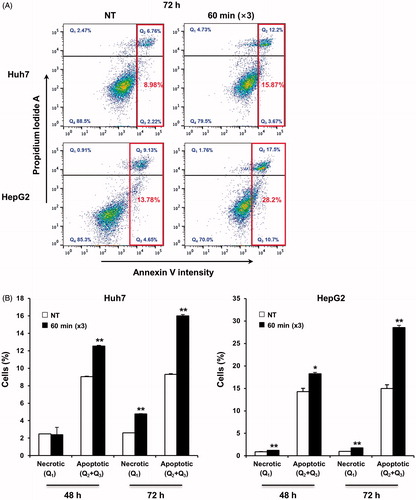
To examine the therapeutic efficacy of mEHT in vitro, two HCC cell lines (Huh7 and HepG2) were treated for 1 h with mEHT three times at 2-day intervals. Cell growth was determined 72 h after the third mEHT using an MTS assay. The temperature in the treatment cup (a cup containing culture medium and a slide plated with cells) increased to 42 °C within approximately 10 min and remained in the range of 42–43 °C (). The mEHT treatments inhibited cell growth by approximately 50% and 53% in the Huh7 and HepG2 cells, respectively (). The decrease in cell viability was affirmed by microscopic observation (). This treatment condition was applied to all subsequent in vitro studies. Consistent with the results of the short-term phenotypic assay, mEHT was also effective in inhibiting long-term colony formation (). Importantly, HCC cells treated with mEHT showed a strong induction of apoptosis, as measured by a concomitant accumulation of apoptosis-specific caspase-cleaved K18 (ccK18) fragments in 48 h of the third mEHT (). We have confirmed the phenotypic change by FACS analysis with Annexin V/PI staining. Overall, mEHT induced both necrosis and apoptosis in HCC cells at 72 h after the third mEHT treatment (). In particular, mEHT significantly increased early (Q3 region) and/or late-apoptotic cell populations (Q2 region) from 8.98% to 15.87% and from 13.78% to 28.2% in Huh7 and HepG2 cells, respectively. However, as shown in , the treatment mainly caused an increase in apoptotic cell population, but not in necrotic cell population in both Huh7 and HepG2 cells at 48 h, the earliest time point, supporting the onset of apoptosis by mEHT. These data show that mEHT inhibits proliferation and induces apoptosis in human HCC cells.
Figure 1. Real-time monitoring of the constant mEHT temperature of 42 °C using sensing probes. Two probes were used to detect the temperature within a treatment cup connected to a LAB-EHY 100 device. Two additional probes were placed outside the treatment cup to detect the temperature of the surroundings.
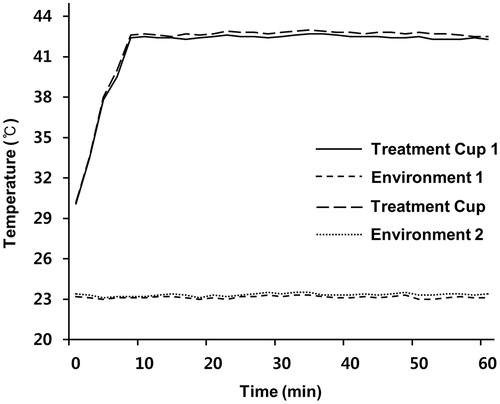
Figure 2. mEHT suppresses proliferation in HCC cells. (A) Measurement of cell viability in Huh7 and HepG2 cells 72 h after the third mEHT treatment. Data represent three independent experiments. NT, no treatment; *P < 0.05 by Wilcoxon rank sum test. (B) Representative light microscopy images of Huh7 and HepG2 cells 72 h after three mEHT treatments. Scale bar, 100 μm.
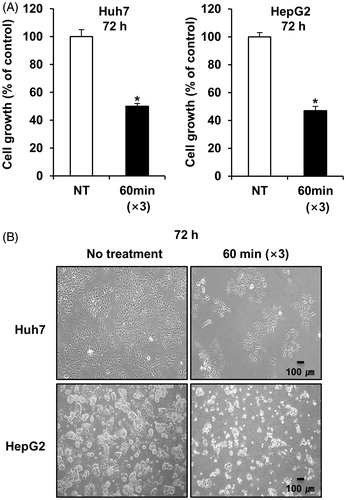
Figure 3. mEHT inhibits long-term colony formation and induces apoptosis in HCC cells. (A) Observation of colony formation using a clonogenic assay 12 days after the third mEHT treatment. Data represent three independent experiments. NT, no treatment; **P < 0.01 by t-test with equal variance. (B) Detection of caspase-cleaved keratin 18 (ccK18) fragments by M30 Apoptosense ELISA 48 h after the third mEHT. Data represent three independent experiments. *P < 0.05 by Wilcoxon rank sum test.
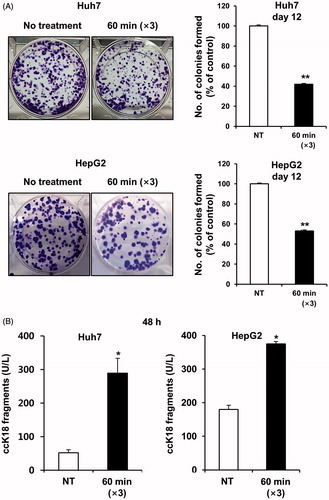
Figure 4. mEHT induces apoptosis in HCC cells. (A) Apoptotic cell population was analysed by FACS by detecting the intensity of apoptotic marker, Annexin V, at 72 h after the third mEHT treatment. Representative results from three independent experiments are shown. (B) Detection of the apoptotic cell population through FACS analysis based on the intensity of the apoptotic marker Annexin V in Huh7 and HepG2 cells at 48 h and 72 h after the third mEHT treatment. The data represent three independent experiments. **P < 0.01, *P < 0.05 by Student’s t-test with equal variance.
The molecular mechanisms underlying the growth inhibitory effects of mEHT in HCC cells
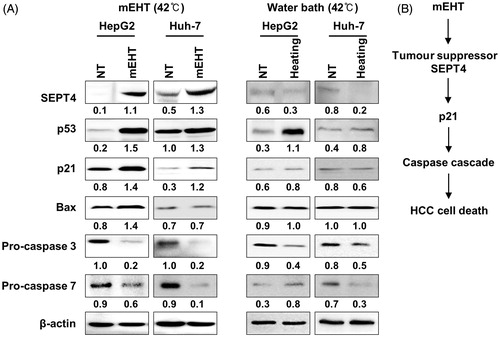
To determine the molecular basis of the observed growth inhibition caused by mEHT, we compared the global gene expression profiles of mEHT-treated Huh7 and HepG2 cells to those of sham-treated control cells using next-generation RNA sequencing. Paired-end mRNA sequencing analysis revealed that mEHT resulted in the up- and down-regulation of 651 RNA transcripts in Huh7 and 2135 transcripts in HepG2 cells (). A comparison of these two gene sets to identify a common anti-HCC mechanism revealed a significant overlap of 37 genes (22 up- and 15 down-regulated genes) defined using a bootstrap ANOVA with 10,000 repetitions and at least a twofold change (P < 0.001) (); this effect was considered to be a common mEHT molecular signature in HCC cells. Ingenuity pathway analysis (IPA) showed that the 37 genes were functionally enriched in the top two networks strongly associated with ubiquitin C (UBC, network 1) and β-catenin (CTNNB1, network 2) (). Consistent with the phenotypic changes, mEHT coordinately impacted a restricted number of defined oncogenic pathways that acted in concert to decrease cell proliferation and induce apoptosis, thereby providing a rationale for the therapeutic use of mEHT in HCC. Specifically, the expression level of SEPT4, which is involved in pro-apoptotic activity and growth suppression [Citation12] was induced, whereas a key regulator of invasiveness and metastasis GPR64 [Citation13] was repressed in RNA level. Subsequent Western blot analysis confirmed the common increase of the SEPT4 tumour suppressor in addition to a concomitant increase in CDK inhibitor p21 and decrease in both pro-caspase 3 and pro-caspase 7 (), thereby accelerating apoptotic signalling in the HepG2 and Huh7 cell lines. The increased level of the p53 tumour suppressor varied according to the different p53 genetic status between the HepG2 (with wild-type (wt) p53) and Huh7 (with Y220C mutant (mt) p53) cell lines. Pro-apoptotic Bax was increased in the HepG2 cells, but not in the Huh7 cells. Next, to investigate whether SEPT4-mediated apoptotic induction was mEHT specific, we assessed the levels of SEPT4, p53, p21, Bax, pro-caspase-3 and pro-caspase-7 in HCC cells by heating three times for 1 h with a two-day interval in a 42 °C water bath, and compared them with mEHT-treated cells. As shown in (right panel), the heat stress itself has up-regulated p53 and down-regulated pro-caspase-3 expression, but SEPT4 and p21 were not up-regulated. This finding suggests that the up-regulation of the tumour suppressor SEPT4 is an essential and common mechanism that suppresses the growth of mEHT-treated HCC cells ().
Figure 5. Transcriptomic analysis of gene expression by RNA sequencing following mEHT treatment. (A) Venn diagrams representing the number of mRNA transcripts up- or down-regulated in Huh7 or HepG2 cells 48 h after the third mEHT. (B) A heat map of 37 commonly deregulated genes in Huh7 and HepG2 cells after normalisation to the corresponding sham-treated cells (10,000 repetitions in bootstrap ANOVA with contrast tests and a threshold cut-off of twofold change, **P < 0.001, log2-based scale). (C, D) Two putative top networks with high scores (>19) strongly associated with ubiquitin C (UBC) (C) and β-catenin (CTNNB1) (D) based on Fisher’s exact test (p value <10−19), indicating a high probability of biological interactions.
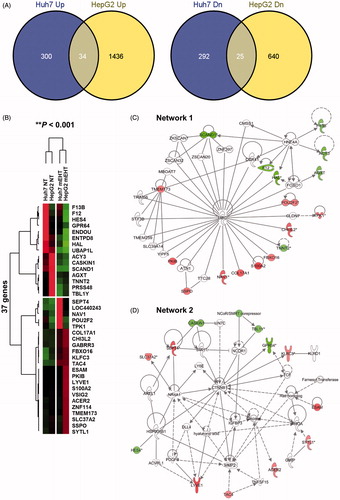
Figure 6. mEHT induces apoptotic signalling through the up-regulation of SEPT4 in HCC cells. (A) Western blot analysis of SEPT4, p53, p21, Bax, pro-caspase 3 and pro-caspase 7, which are functionally involved in cell growth or apoptotic progression. Whole cell lysates were prepared 24 h after the third mEHT treatment or water bath heating. β-actin was included as a loading control. NT, no treatment. The intensity of each band was quantified by densitometry using Image J software. Relative band intensities between the control and the mEHT treatment are numerically shown after normalising to β-actin protein expression. (B) Schematic diagram of mEHT-induced up-regulation of SEPT4 and p21 that is responsible for HCC cell death.
mEHT suppresses HCC growth in vivo
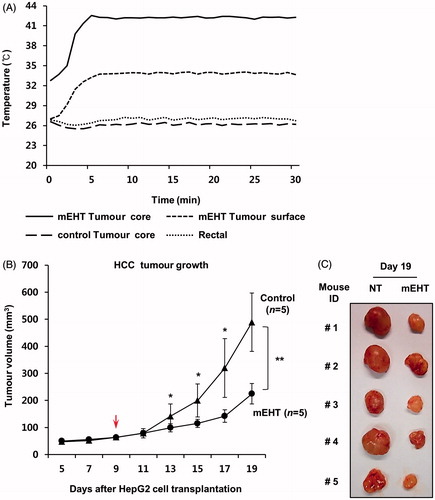
We used HepG2 xenografts in nude mice to test the therapeutic efficacy of mEHT in vivo. mEHT was directly applied five times to the surface of each tumour on days 9, 11, 13, 15, and 17 after HepG2 cell transplantation. As shown in , the temperature within the tumour core increased to 42 °C within 5 min and remained in the range of 42–43 °C during heating using the LAB-EHY100 device to control RF power. Tumour growth was monitored using a caliper ruler. As a result, the mEHT treatments significantly suppressed the growth of human HCC xenografts (). On day 19, the mean volume to tumours treated with mEHT was 200 mm3, whereas that of control tumours was 490 mm3 (*P < 0.05).
Figure 7. mEHT inhibits the growth of human HCC xenograft in mice. (A) Real-time monitoring of the constant temperature of 42 °C in the tumour core using the sensing probes. Two probes were used to detect the temperature within a tumour core or a tumour surface. Another two probes were used to monitor the real-time temperature within a control tumour (not mEHT-treated) and the rectum. (B) Kinetics of tumour growth: mEHT was directly applied to the surface of each tumour beginning 9 days after inoculation of 1 × 107 HepG2 cells. Tumour diameters were measured on the indicated days with digital calipers. The red arrow indicates the first day of mEHT treatment. The results are shown as the mean tumour volume ± SD. **P < 0.01, *P < 0.05 (n = 5 versus n = 5) by normality assumption test. (C) Gross tumour morphology 19 days after HepG2 cell transplantation.
Discussion
mEHT is a locoregional hyperthermia method that is widely used in combination with chemotherapy or radiation therapy. mEHT concentrates the modulated radiofrequency current to intercellular electrolytes and generates a temperature gradient, thereby destroying the tumour cell membrane and leading to apoptosis or necrosis [Citation9,Citation10]. Many clinical studies have shown that mEHT is effective for controlling advanced primary and metastatic malignancies in liver cancer, bone (metastatic) cancer, breast cancer and malignant glioma [Citation11,Citation14–17]. Yet the molecular mechanism underlying the induction of tumour cell death by mEHT remains unclear.
In this study we observed that mEHT induced extensive apoptosis in both HepG2 and Huh7 HCC cell lines, as evidenced by the detection of ccK18 fragments () and apoptotic cell population (). This phenomenon was due to the coordinated and common dysregulation of 37 genes (), as assessed through a global transcriptomic analysis using RNA sequencing. Representatively, mEHT-induced molecular alterations were associated with the increased expression of the proapoptotic effector SEPT4 and the down-regulation of key regulators of cell growth (i.e. GRP64) in RNA level. As shown in , it was demonstrated that mEHT generally increased the protein expressions of SEPT4 and p21 proteins to mediate apoptosis in HCC cells, and these molecular alterations were mEHT specific. This observation is in agreement with recent findings that SEPT4, which encodes the IAP (inhibitor of apoptosis protein) antagonist ARTS, is significantly down-regulated in human HCC cells and regulates susceptibility to apoptosis, suggesting a tumour-suppressor role of SEPT4 in HCC [Citation12]. Sept4-null mice had increased numbers of stem and progenitor cells, elevated X-linked IAP (XIAP) protein, increased resistance to cell death, and accelerated tumour development in a Myc-dependent manner [Citation18,Citation19], and synthetic peptides mimicking the unique ARTS-XIAP binding site promoted apoptosis in cultured cancer cells [Citation20]. Notably, mEHT treatment commonly induced p21 levels in both HepG2 (with wt p53) and Huh7 cells (with Y220C mt p53), suggesting that the mEHT may be applied to HCC patients regardless of their p53 genetic status. Pro-apoptotic Bax was increased in HepG2 cells but not in Huh7 cells, showing a cell-line dependency. This result could be partially explained by the previous observation that p53 mutants can often transactivate the p21 promoter, but not Bax [Citation21]. However, the mechanism by which p21 but not Bax was induced in cancer cells with mt p53 is still unknown. In future studies further investigations into the primary effect of mEHT that can elicit the growth inhibition of HCC cells with p53 mutations is warranted. The activation of the tumour-suppressive SEPT4 genes and subsequent induction of p21 by mEHT might represent a common molecular mechanism to mediate growth inhibition and apoptotic induction in HCC cells with different p53 statuses.
In conclusion, in the present study using human HCC cell lines we found out that mEHT inhibited cell proliferation and clonogenicity, induced apoptosis in vitro, and suppressed the growth of a HCC xenograft in nude mice. We also provided evidence for the molecular mechanism by which mEHT induces apoptosis in HCC cells. The antitumorigenic effects of mEHT might reflect the up-regulation of tumour suppressor SEPT4 that commonly activates p21-dependent apoptotic signalling.
Acknowledgement
*T.W.J. and H.Y. contributed equally to this work.
Disclosure statement
This project was financially supported through the Hospicare Corp. (2013-31-0557), South Korea, and through grants from the Korean Health Technology R&D Project, Ministry of Health and Welfare (A121982) and the Bio and Medical Technology Development Programme, Ministry of Science, ICT and Future Planning (NRF-2012M3A9C7050149). This work was also supported by the Mid-Career Researcher Programme through a National Research Foundation grant funded by the Ministry of Education, Science and Technology (NRF-2014R1A2A1A11054162), South Korea. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Related Research Data
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61:69–90.
- Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol 2010;7:448–58.
- Marquardt JU, Thorgeirsson SS. Snapshot: hepatocellular carcinoma. Cancer Cell 2014;25:550.e1.
- Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet 2012;379(9822):1245–55.
- Hildebrandt B, Wust P, Ahlers O, et al. The cellular and molecular basis of hyperthermia. Crit Rev Oncol Hematol 2002;43:33–56.
- Taraya-Brown S, Fiering S. Local tumour hyperthermia as immunotherapy for metastatic cancer. Int J Hyperthermia 2014;30:531–9.
- Andocs G, Szasz O, Szasz A. Oncothermia treatment of cancer: from the laboratory to clinic. Electromagn Biol Med 2009;28:148–65.
- Hegyi G, Szigeti GP, Szasz A. Hyperthermia versus oncothermia: cellular effects in complementary cancer therapy. Evid Based Complement Alternat Med 2013;2013:672873.
- Szasz O. Essentials of oncothermia. Conf Papers Med 2013;159570:1–20.
- Szasz A. Electromagnetic effects of nanoscale range. In: Shimizu T, Kondo T, eds. Cellular response to physical stress and therapeutic application. New York: Nova Biomedical; 2013.
- Gadaleta-Caldarola G, Infusino S, Galise I, Ranieri G, Vinciarelli G, Fazio V, et al. Sorafenib and locoregional deep electro-hyperthermia in advanced hepatocellular carcinoma: a phase II study. Oncol Lett 2014;8:1783–7.
- Shen S, Liu M, Wu Y, Saiyin H, Liu G, Yu L. Involvement of SEPT4_i1 in hepatocellular carcinoma: SEPT4_i1 regulates susceptibility to apoptosis in hepatocellular carcinoma cells. Mol Biol Rep 2012;39:4519–26.
- Richter GH, Fasan A, Hauer K, Grunewald TG, Berns C, Rössler S, et al. G-protein coupled receptor 64 promotes invasiveness and metastasis in Ewing sarcomas through PGF and MMP1. J Pathol 2013;230:70–81.
- Bogovic J, Douwes F, Muravjov G, Istomin J. Posttreatment histology and microcirculation status of osteogenic sarcoma after a neoadjuvant chemo- and radiotherapy in combination with local electromagnetic hyperthermia. Onkologie 2001;24:55–8.
- Feyerabend T, Wiedemann GJ, Jager B, Vesely H, Mahlmann B, Richter E. Local hyperthermia, radiation, and chemotherapy in recurrent breast cancer is feasible and effective except for inflammatory disease. Int J Radiat Oncol Biol Phys 2001;49:1317–25.
- Fiorentini G, Giovanis P, Rossi S, Dentico P, Paola R, Turrisi G, et al. A phase II clinical study on relapsed malignant gliomas treated with electro-hyperthermia. In Vivo 2006;20:721–4.
- Fiorentini G, Szasz A. Hyperthermia today: electric energy, a new opportunity in cancer treatment. J Cancer Res Ther 2006;2:41–6.
- García-Fernández M, Kissel H, Brown S, Gorenc T, Schile AJ, Rafii S, et al. Sept4/ARTS is required for stem cell apoptosis and tumor suppression. Genes Dev 2010;24:2282–93.
- Fuchs Y, Brown S, Gorenc T, Rodriguez J, Fuchs E, Steller H. Sept4/ARTS regulates stem cell apoptosis and skin regeneration. Science 2013;341:286–9.
- Edison N, Reingewertz TH, Gottfried Y, Lev T, Zuri D, Maniv I, et al. Peptides mimicking the unique ARTS-XIAP binding site promote apoptotic cell death in cultured cancer cells. Clin Cancer Res 2012;18:2569–78.
- Campomenosi P, Monti P, Aprile A, Abbondandolo A, Frebourg T, Gold B, et al. p53 mutants can often transactivate promoters containing a p21 but not Bax or PIG3 responsive elements. Oncogene 2001;20:3573–9.