
Abstract
Purpose: Heat shock induces DNA double-strand breaks (DSBs) in mammalian cells. Mammalian cells are capable of repairing DSBs by utilising the homologous recombination (HR) pathway. Breast cancer susceptibility gene 2 (BRCA2) is known to regulate the HR pathway. Here, we investigate the role of BRCA2 in repairing DNA damage induced by heat shock.
Materials and methods: Chinese hamster lung fibroblast cell lines and human tongue squamous cell carcinoma SAS cells were used. RAD51 foci formation assay was used as an HR indicator. Heat sensitivity was analysed with colony forming assays. Phosphorylated histone H2AX (γH2AX) intensity, which correlates with the number of DSBs, was analysed with flow cytometry.
Results: RAD51 foci appeared with heat shock, and the number of cells with RAD51 foci was maximal at about 4 h after heat shock. Heat-induced RAD51 foci co-localised with γH2AX foci. BRCA2-deficient cells were sensitive to heat when compared to their parental wild-type cells. Heat-induced γH2AX was higher in BRCA2-deficient cells compared to parental cells. In SAS cells, cells transfected with BRCA2-siRNA were more sensitive to heat than cells transfected with negative control siRNA. Apoptotic bodies increased in number more rapidly in BRCA2-siRNA transfected cells than in cells transfected with negative control siRNA when cells were observed at 48 h after a heat treatment. In addition, cells deficient in BRCA2 were incapable of activating heat-induced G2/M arrest.
Conclusion: BRCA2 has a protecting role against heat-induced cell death. BRCA2 might be a potential molecular target for hyperthermic cancer therapy.
Introduction
Hyperthermia is currently used to treat various types of cancer. Heat shock induces DNA double-strand breaks (DSBs) in mammalian cells [Citation1,Citation2], and unrepaired DSBs are the most lethal DNA damage [Citation3]; however, the molecular mechanism of cellular response to heat-induced DNA damage is not fully understood.
Two pathways are known to be involved in DSB repair in mammalian cells: non-homologous end-joining (NHEJ) repair and homologous recombination (HR) repair. NHEJ repair is active throughout all cell cycle phases, and is mediated by DNA-dependent protein kinase [DNA-PK: DNA-PKcs (catalytic subunit) and Ku70/80] [Citation4]. HR repair takes advantage of either of the homologous chromosomes or the sister chromatid to correctly re-join broken DNA ends during late S and G2 phases, but not during G1 phase [Citation5]. Proteins involved in the HR repair pathway in vertebrate cells include BRCA2 (breast cancer susceptibility gene 2), RAD52, RAD54 and RAD51 paralogs such as X-ray repair cross-complementing protein 2/3 (XRCC2/3), RAD51B, RAD51C and RAD51D [Citation6]. RAD51 activity is regulated by BRCA2, which is an upstream-protein to RAD51 [Citation7]. We recently reported that mammalian cells are capable of repairing DSBs by utilising the HR pathway [Citation8]. However, the precise mechanism as to how DNA repair is processed under heat shock remains unclear.
It has been agreed upon that hyperthermia inhibits various DNA repair pathways, including the NHEJ and HR pathways [Citation9]. The exact mechanisms by which hyperthermia inhibits NHEJ and HR have not been elucidated, but with NHEJ, it has been seen that heat shock lowers activity levels of DNA-PKcs [Citation10–12] as well as protein levels of Ku70/80 [Citation13,Citation14], overall preventing proper functioning of the NHEJ pathway. In addition, BRCA2 is degraded by heat shock [Citation15–20], likely leading to inhibition of HR; thus hyperthermia inhibits DNA repair for at least a certain period of time.
In this study, we report that heat shock induced RAD51 foci and that BRCA2 plays a protective role in heat-induced cell death by repairing heat-induced DSBs. These findings provide an insight into better understanding of how heat-induced DNA damage is repaired.
Materials and methods
Cell culture
The Chinese hamster (CH) lung fibroblast cell lines used in this study were V79 (BRCA2-wild type), V-C8 (BRCA2-deficient), V-C8 + BRCA2 (BRCA2-revertant, V-C8 containing a BAC with the murine BRCA2 gene) [Citation21] (kindly provided by Dr. M.Z. Zdzienicka), V79B (Ku80-wild type) and XR-V15B (Ku80-deficient cell line) (kindly provided by Dr. Akira Yasui). The human cell line was a tongue squamous cell carcinoma cell (SAS). Cells were cultured at 37 °C in Dulbecco's modified Eagle’s medium containing 10% (v/v) foetal bovine serum, penicillin (50 U/ml), streptomycin (50 µg/ml) and kanamycin (50 µg/ml) (DMEM-10).
Heat treatment and irradiation
Exponentially growing cells were immersed in a water bath (Thermominder EX, Taitec Co. Ltd., Saitama, Japan) maintained at 44 ± 0.1 °C. X-irradiation (1.0 Gy/min) was administered with a 150-kVp X-ray generator (Model MBR-1520R; Hitachi, Tokyo, Japan). Under the present experimental conditions, no marked change in pH values was detected in the medium during the treatments.
Colony forming assays
Cell survival was measured using a standard colony forming assay, as previously described [Citation22]. Three flasks were used for each point, and three independent experiments were repeated for each point. Colonies obtained after 7–10 days were fixed with methanol and stained with a 2% Giemsa solution. Microscopic colonies composed of more than approximately 50 cells were counted as having arisen from single surviving cells. The sensitivity of each cell line was assessed by its D50 or T50 value, that is, from the radiation dose or the heating period which reduced cell survival to 50%. In order to accurately compare sensitivities to radiation or heat in the repair defective cell lines, the relative D50 or T50 values were normalised using the D50 or T50 value of the parental cell lines.
Interference RNA
The siRNA sequence used for human BRCA2 was AAC AAC AAU UAG GAA CCA AAC UU [Citation23]. The siRNA sequence of the non-specific negative control was the same as used previously [Citation24]. The siRNA duplexes were synthesised and provided as a purified and annealed duplex by the Japan Bio Services Co., Ltd. (Saitama, Japan). Human BRCA2 siRNA or a non-specific negative control siRNA was transfected into human tongue squamous cell carcinoma SAS cells as previously described [Citation22]. The siRNA sequences targeting BRCA2 used here are the most commonly used in other reported work. Transfections were performed using Lipofectamine RNAiMAX. Cells were seeded at 1–5 × 104 cells per 6 cm plate for 16–24 h without antibiotics. The siRNA was diluted in Opti-MEM I (Invitrogen, Carlsbad, CA) to produce a final siRNA concentration of 10 nM in a 1 ml final transfection volume. In a separate tube, 10 μl of Lipofectamine RNAiMAX was added to 490 μl of Opti-MEM I. The Lipofectamine RNAiMAX dilution was mixed with the diluted siRNA and incubated at room temperature for 15 min. The complex was then added drop-wise onto the cells. The cells were incubated for 48 h before further processing. The cells were then trypsinised and used for colony forming assays or western blot analysis.
Western blots
Total cellular protein was determined with a Bio-Rad protein assay kit (Bio-Rad Labs, Richmond, CA). Aliquots of proteins (20 μg) were subjected to Western blot analysis. Total cell lysates were loaded onto 7% Tris-glycine gels (Invitrogen, Carlsbad, CA), separated by electrophoresis at a constant voltage (125 V) and electro-transferred onto nitrocellulose membranes at 42 V. Membranes were blocked for 1 h at room temperature in blocking buffer (25 mM Tris pH 8.0, 125 mM NaCl, 1% Tween 20 [TBS-T buffer] containing 5% skim milk) and incubated with mouse monoclonal anti-BRCA2 antibodies (Ab-4, Calbiochem and Oncogene, Calbiochem, Germany) or goat polyclonal anti-actin antibodies (I-19; Santa Cruz Biotechnology, Santa Cruz, CA), the primary antibodies, for 2 h at room temperature. The membranes were washed with TBS-T buffer three times and incubated with a secondary antibody conjugated to horseradish peroxidase for 1 h. After washing three times, blots were visualised by scanning profiles using the Scion imaging programme (Scion, Frederick, MD). The relative ratio of the two bands was used to measure the level of BRCA2 protein expression, and the values used were averages from three experiments. Measurements were obtained by densitometry following β-actin normalisation with and without BRCA2 siRNA.
Analysis of apoptosis
Induction of apoptosis was analysed by detecting apoptotic bodies with a Hoechst33342 staining assay. Cells were fixed with 1% glutaraldehyde (Nacalai Tesque, Kyoto, Japan) in PBS at 4 °C, washed with PBS, stained with 0.2 mM Hoechst33342 (Nacalai Tesque) and then observed under a fluorescence microscope.
Immunocytochemistry
Cells were grown on cover slips in a 30 mm dish, fixed in 2% paraformaldehyde in PBS for 15 min at room temperature and washed in PBS. Then the cells were permeabilised for 5 min at 4 °C in 0.2% Triton X-100, and blocked in PBS with 1% Bovine Serum Albumin (BSA) for 1 h at 37 °C. Cells were then incubated with anti-phospho-H2AX (Ser 139) mouse monoclonal antibodies (Upstate Biotechnology, Lake Placid, NY) at a 1:300 dilution, or rabbit polyclonal anti-RAD51 primary antibodies (H-92, Santa Cruz) for 1 h at room temperature at a 1:300 dilution in PBS containing 1% BSA. Cells were then washed three times in PBS containing 1% BSA for 10 min. The cells were incubated with an Alexa Fluor 488-conjugated secondary antibody (Molecular Probes, Eugene, OR) for 1 h at room temperature at a 1:400 dilution in PBS containing 1% BSA, and washed three times for 10 min in PBS. Cover slips were mounted with a 1:1000 dilution of 4,6-diamidino-2-phenylindole (DAPI). Fluorescence images were captured using a fluorescence microscope (Keyence, Tokyo, Japan) for analysis.
H2AX phosphorylation analysis by flow cytometry
After treatment, cells were fixed in cold 70% methanol and kept at 4 °C for up to 1 week before analysis. Cells were centrifuged and rinsed with PBS with 0.1% Tween 20 (TPBS). The cells were blocked with bovine serum for 15 min at room temperature and rinsed with TPBS. Cells were then incubated with anti-phospho-H2AX (Ser 139) monoclonal antibody (JBW301; Millipore, Billerica, MA) at a 300-fold dilution for 60 min at room temperature, rinsed with TPBS, incubated with an AlexaFluor 488-conjugated anti-mouse IgG secondary antibody (Invitrogen) at a 400-fold dilution for 60 min at room temperature, and then rinsed in TPBS. Before flow cytometric analysis, samples were filtered through a 35 μm nylon mesh. Samples were analysed using a flow cytometer (Becton Dickinson, San Jose, CA). The γH2AX levels at 0.5 h were defined as 100%, and the γH2AX levels at 18 h after heat treatment were determined and are shown as a percentage of the values at 0.5 h.
Cell cycle analysis
After treatment, cells were fixed with cold 70% methanol and stored at 4 °C for 3 days before analysis. For cell cycle analysis, the cells were incubated for 30 min at room temperature with 1 mg/mL RNase and 50 μg/mL propidium iodide (PI), and were analysed using a flow cytometer. The cell cycle distribution was measured by determining the DNA content twice and calculating the average.
Statistical analysis
Statistical analysis was performed using Student’s t-test.
Results
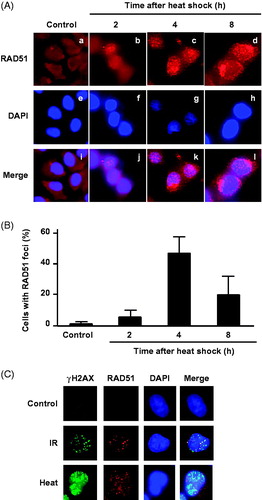
The BRCA2-mediated DSB repair pathway is followed by HR, and RAD51 foci formation is commonly used as an indicator of HR repair. To verify the role of HR in heat-induced DSB repair, we examined RAD51 foci formation after heat treatment. RAD51 foci were observed in the nuclei of V79 (parental) cells after heat treatment at 44 °C for 60 min (). RAD51 foci number reached a peak at 4 h after heat treatment (). Moreover, at 4 h after heat-treatment (44 °C for 60 min), RAD51 co-localised with γH2AX foci in the nucleus ().
Figure 1. RAD51 foci formation after heat shock. (A) Typical photographs of RAD51 in V79 cells at indicated time points after heat shock (44 °C for 60 min). a–d, RAD51; e–h, DAPI; i–l, merge. (B) RAD51 foci formation at indicated times after heat shock. Columns show the mean of three independent experiments. Bars indicate the SD. (C) Immunofluorescence staining for γH2AX and RAD51 foci formation in nuclei by DAPI of V79 cells at 4 h after heat shock (44 °C for 60 min) and irradiation (X-rays, 10 Gy).
To verify the cell lines used in this study, we first confirmed cellular sensitivity to X-rays by using Ku80-deficient cells, BRCA2-deficient cells and their parental CH cells. Both types of cells were more sensitive to X-rays than their corresponding parental cells (). Each D50 value was calculated from the cell survival data shown in . The relative D50 values are listed sequentially in the order in which they increase (reflecting a decreasing sensitivity to X-rays): Ku80-deficient cells (14%) < BRCA2-deficient cells (25%) < BRCA2-revertant cells (75%) ().
Figure 2. BRCA2-deficient cells are sensitive to heat shock. (A) X-irradiation, (B) heat shock. Left panel, Ku80-wild type cells (open circles), Ku80-deficient cells (closed circles); Right panel, BRCA2-wild type cells (open circles), BRCA2-deficient cells (closed circles), BRCA2-revertant cells (triangles). Each point represents the mean of three independent experiments; bars indicate the SD. (C) relative D50 values for X-ray sensitivity. (D) Relative T50 values for heat sensitivity. (E) Relative induced γH2AX levels at different time points were normalised against the γH2AX levels measured at 0.5 and 18 h after heat treatment (44 °C for 20 min) with flow cytometry analysis. The γH2AX levels at 0.5 h were set to 100%, and relative γH2AX levels at 18 h were calculated and are shown as percentages in the graph. Closed columns, BRCA2-deficient cells; open columns, the parental cells. The columns show the mean of at least three independent experiments; vertical bars indicate the SD.
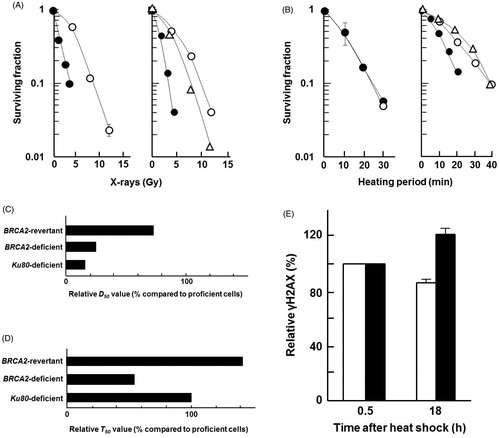
To verify whether the NHEJ or HR repair pathway (especially BRCA2-mediated) plays a more important role in survival from heat-induced cell death, cellular responses to heat were examined using Ku80-deficient cells, BRCA2-deficient cells and the parental cells. There was no difference in the heat sensitivity of Ku80-deficient cells and their parental cells; however, BRCA2-deficient cells were more sensitive to heat than their parental cells (). The sensitivity of each cell line was assessed from its T50 value in . The relative T50 values are listed sequentially in the order in which they increase (reflecting decreasing sensitivities to heat): BRCA2-deficient cells (55%) < Ku80-deficient cells (100%) < BRCA2-revertant cells (144%) (). Heat-induced γH2AX was higher in BRCA2-deficient cells (122%) compared to parental cells (85%) at 18 h after heat shock (44 °C for 20 min). Together, these results suggest that BRCA2 has a protective role by repairing heat-induced DSBs.
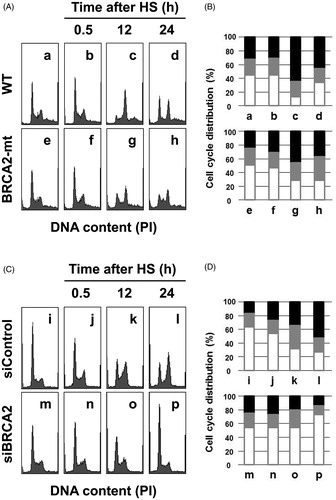
Next, we examined heat sensitivity using human tongue squamous cell carcinoma SAS cells by knocking down BRCA2. BRCA2 knockdown was confirmed by western blotting (). Cells with siBRCA2 were more sensitive to heat treatment than control (). The relative T50 values of BRCA2 siRNA transfected SAS cells were 62%. Apoptotic bodies appeared at a higher frequency in the cells with siBRCA2 (). At 48 h after heat treatment, apoptosis frequencies in the BRCA2-siRNA transfected cells and the negative control-siRNA transfected cells were 27% and 14%, respectively (). We then examined the contribution of BRCA2 in cell cycle distribution after heat treatment. Both in CH and SAS cells, BRCA2 deficiency led to a lower level of cells accumulated at G2/M phases (), indicating that BRCA2 is important for G2/M arrest after heat shock.
Figure 3. Knocking down BRCA2 enhanced sensitivity to heat shock. (A) Expression analysis by western blot of BRCA2 in SAS cells transfected with siRNA targeting human BRCA2 or a negative control siRNA. Actin was used as a loading control, and the relative ratios of protein were normalised using actin levels. (B) Effect of siRNA silencing of BRCA2 on cellular sensitivity to heat treatment; SAS cells transfected with siRNA for BRCA2 (closed circles), SAS cells transfected with the negative control siRNA (open circles). (C) Typical photographs of the BRCA2-siRNA transfected cells; a, untreated control cells; b, 48 h after heat shock at 44 °C for 20 min. Arrows indicate typical apoptotic bodies. (D) Time-dependent apoptosis rates up to 48 h after heat shock (44 °C for 20 min).
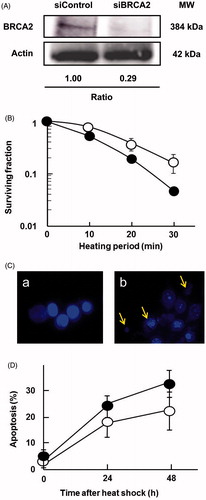
Figure 4. Cell cycle analysis after heat shock. (A) and (B) Cell cycle histograms with flow cytometry and cell cycle distributions from histogram analysis in Chinese hamster cells; a–d, wild-type cells; e–h, BRCA2-deficient cells. a and e, untreated controls; b and f, 0.5 h; c and g, 12 h; d and h, 24 h after heat shock (HS) at 44 °C for 20 min. (C) and (D) Cell cycle histograms with flow cytometry and cell cycle distributions from histogram analysis in human SAS cells; i–l, negative control-siRNA transfected cells; m–p, BRCA2-siRNA transfected cells. i and m, untreated controls; j and n, 0.5 h; k and o, 24 h; l and p, 24 h after heat shock at 44 °C for 30 min. Open columns, G1; grey columns, S; closed columns, G2/M.
Discussion
Our recent work revealed that the HR pathway, but not NHEJ, plays an important role in the survival of mammalian cells against cell death induced by heat shock. Here in this study, we further investigated the role of BRCA2, upstream factors important for the HR pathway. Heat shock caused RAD51 foci formation, which has been extensively used as an indicator of HR repair, and those RAD51 foci were co-localised with γH2AX foci after heat shock (). BRCA2-deficient cells (CH cells and human cancer cells) were sensitive to heat shock and had more remaining γH2AX signal ().
The study by Oei et al. [Citation19] compared the effects of heat shock on the sensitivity of BRCA2-deficient and proficient cells, though at 42 °C and for 1 h along with exposure to radiotherapy. With the different experimental conditions, it is difficult to directly compare the results of that study with ours (experiments with heat shock alone). It seems that while our study can generally conclude that BRCA2-deficient cells are more sensitive than BRCA2-proficient cells, Oei et al. did not produce as clear of a distinction between sensitivities of BRCA2-deficient and proficient cells, and rather focussed on differences in sensitivity between treatment groups (radiation with and without heat treatment).
BRCA2 has multiple roles in maintaining genomic stability, dependent and independent of DSB repair [Citation25]. BRCA2 is required for survival in the presence of 5-fluorouracil [Citation22], formaldehyde [Citation26] and DNA alkylating agents [Citation24]. In this study, we have shown that BRCA2 has a protective role in sensitivity to heat shock and DSB repair. The results suggest that in response to heat shock, BRCA2, at least, acts as an HR repair factor to repair DSBs caused by heat shock, although the possibility of other roles of BRCA2 still remain unclear.
Heat shock affects stability/activity of several DNA repair factors: Ku70/80 [Citation13,Citation14], DNA-PK [Citation10–12], MRE11/RAD50/NBS1 [Citation27–29], BRCA1 [Citation30], BRCA2 [15–20] and other HR factors [Citation31]. The NHEJ pathway was shown not to be involved in radio-sensitisation by heat [Citation31–33]. More recently, however another group reported that targeting DNA-PKcs enhanced radio-sensitisation by heat [Citation34]. This discrepancy between different groups may come from research materials they used (i.e. cell type, species). On the other hand, the HR pathway is a potential target for thermal-radio-sensitisation [Citation15,Citation31]. Heat shock has two effects on BRCA2: (1) protein degradation [Citation15] and (2) repairing DSBs induced by heat shock (this study). The simplest interpretation is that heat shock leads to degradation of BRCA2, but enough BRCA2 remains to participate in the HR and repair DSBs induced by heat shock, and cells with insufficient BRCA2 are not capable of repairing DSBs, so they cannot survive. It is also possible that surviving cells are able to translate new BRCA2 with the help of HSP90, a chaperone protein that helps proteins to fold even under high heat and that is known to interact with BRCA2 [Citation35], and the newly translated BRCA2 engages in DSB repair. These reasons may explain why the difference in cellular sensitivity to heat shock is not as large as radiation sensitivity in NHEJ deficiency.
Taken together, BRCA2 has the potential to be a useful molecular target during hyperthermia therapies. Simultaneously down-regulating the BRCA2 gene in conjunction with hyperthermia treatments could theoretically provide a protocol with the potential to enhance hyperthermia results in cancer patients.
Acknowledgements
The authors thank Dr. M.Z. Zdzienicka and Dr. A. Yasui for kindly providing cell lines used in this work.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Takahashi A, Matsumoto H, Nagayama K, et al. (2004). Evidence for the involvement of double-strand breaks in heat-induced cell killing. Cancer Res 64:8839–45.
- Takahashi A, Mori E, Somakos GI, et al. (2008). Heat induces γH2AX foci formation in mammalian cells. Mutat Res 656:88–92.
- Ohnishi T, Mori E, Takahashi A. (2009). DNA double-strand breaks: their production, recognition, and repair in eukaryotes. Mutat Res 669:8–12.
- Davis AJ, Chen DJ. (2013). DNA double strand break repair via non-homologous end-joining. Transl Cancer Res 2:130–43.
- Chapman JR, Taylor MR, Boulton SJ. (2012). Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 47:497–510.
- Prakash R, Zhang Y, Feng W, Jasin M. (2015). Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol 7:a016600.
- Davies AA, Masson JY, McIlwraith MJ, et al. (2001). Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol Cell 7:273–82.
- Takahashi A, Mori E, Nakagawa Y, et al. (2017). Homologous recombination preferentially repairs heat-induced DNA double-strand breaks in mammalian cells. Int J Hyperthermia 33:336–42.
- Oei AL, Vriend LE, Crezee J, et al. (2015). Effects of hyperthermia on DNA repair pathways: one treatment to inhibit them all. Radiat Oncol 10:165.
- Ihara M, Suwa A, Komatsu K, et al. (1999). Heat sensitivity of double-stranded DNA-dependent protein kinase (DNA-PK) activity. Int J Radiat Biol 75:253–8.
- Woudstra EC, Konings AW, Jeggo PA, Kampinga HH. (1999). Role of DNA-PK subunits in radiosensitization by hyperthermia. Radiat Res 152:214–18.
- Ihara M, Takeshita S, Okaichi K, et al. (2014). Heat exposure enhances radiosensitivity by depressing DNA-PK kinase activity during double strand break repair. Int J Hyperthermia 30:102–9.
- Burgman P, Ouyang H, Peterson S, et al. (1997). Heat inactivation of Ku autoantigen: possible role in hyperthermic radiosensitization. Cancer Res 57:2847–50.
- Beck BD, Dynlacht JR. (2001). Heat-induced aggregation of XRCC5 (Ku80) in nontolerant and thermotolerant cells. Radiat Res 156:767–74.
- Krawczyk PM, Eppink B, Essers J, et al. (2011). Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proc Natl Acad Sci USA 108:9851–6.
- Bergs JW, Oei AL, Ten Cate R, et al. (2016). Dynamics of chromosomal aberrations, induction of apoptosis, BRCA2 degradation and sensitization to radiation by hyperthermia. Int J Mol Med 38:243–50.
- Harnicek D, Kampmann E, Lauber K, et al. (2016). Hyperthermia adds to trabectedin effectiveness and thermal enhancement is associated with BRCA2 degradation and impairment of DNA homologous recombination repair. Int J Cancer 139:467–79.
- van den Tempel N, Laffeber C, Odijk H, et al. (2017). The effect of thermal dose on hyperthermia-mediated inhibition of DNA repair through homologous recombination. Oncotarget 8:44593–604.
- Oei AL, Ahire VR, van Leeuwen CM, et al. (2017). Enhancing radiosensitisation of BRCA2-proficient and BRCA2-deficient cell lines with hyperthermia and PARP1-i. Int J Hyperthermia. [Epub ahead of print]. DOI: 10.1080/02656736.2017.1324642.
- van den Tempel N, Odijk H, van Holthe N, et al. (2017). Heat-induced BRCA2 degradation in human tumours provides rationale for hyperthermia-PARP-inhibitor combination therapies. Int J Hyperthermia. [Epub ahead of print]. DOI: 10.1080/02656736.2017.1355487.
- Wiegant WW, Overmeer RM, Godthelp BC, et al. (2006). Chinese hamster cell mutant, V-C8, a model for analysis of Brca2 function. Mutat Res 600:79–88.
- Nakagawa Y, Kajihara A, Takahashi A, et al. (2014). The BRCA2 gene is a potential molecular target during 5-fluorouracil therapy in human oral cancer cells. Oncol Rep 31:2001–6.
- Bruun D, Folias A, Akkari Y, et al. (2003). siRNA depletion of BRCA1, but not BRCA2, causes increased genome instability in Fanconi anemia cells. DNA Repair (Amst) 2:1007–13.
- Kondo N, Takahashi A, Mori E, et al. (2011). FANCD1/BRCA2 plays predominant role in the repair of DNA damage induced by ACNU or TMZ. PLoS One 6:e19659.
- Schlacher K, Christ N, Siaud N, et al. (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145:529–42.
- Noda T, Takahashi A, Kondo N, et al. (2011). Repair pathways independent of the Fanconi anemia nuclear core complex play a predominant role in mitigating formaldehyde-induced DNA damage. Biochem Biophys Res Commun 404:206–10.
- Seno JD, Dynlacht JR. (2004). Intracellular redistribution and modification of proteins of the Mre11/Rad50/Nbs1 DNA repair complex following irradiation and heat-shock. J Cell Physiol 199:157–70.
- Zhu WG, Seno JD, Beck BD, Dynlacht JR. (2001). Translocation of MRE11 from the nucleus to the cytoplasm as a mechanism of radiosensitization by heat. Radiat Res 156:95–102.
- Takahashi A, Mori E, Ohnishi T. (2010). The foci of DNA double strand break-recognition proteins localize with γH2AX after heat treatment. J Radiat Res 51:91–5.
- Xian Ma Y, Fan S, Xiong J, et al. (2003). Role of BRCA1 in heat shock response. Oncogene 22:10–27.
- Genet SC, Fujii Y, Maeda J, et al. (2013). Hyperthermia inhibits homologous recombination repair and sensitizes cells to ionizing radiation in a time- and temperature-dependent manner. J Cell Physiol 228:1473–81.
- Iliakis G, Seaner R. (1990). A DNA double-strand break repair-deficient mutant of CHO cells shows reduced radiosensitization after exposure to hyperthermic temperatures in the plateau phase of growth. Int J Hyperthermia 6:801–12.
- Dynlacht JR, Bittner ME, Bethel JA, Beck BD. (2003). The non-homologous end-joining pathway is not involved in the radiosensitization of mammalian cells by heat shock. J Cell Physiol 196:557–64.
- van Oorschot B, Granata G, Di Franco S, et al. (2016). Targeting DNA double strand break repair with hyperthermia and DNA-PKcs inhibition to enhance the effect of radiation treatment. Oncotarget 7:65504–13.
- Pennisi R, Ascenzi P, di Masi A. (2015). Hsp90: a new player in DNA repair? Biomolecules 5:2589–618.