
Abstract
Hyperthermia in pigs induces suppressor of cytokine signaling (SOCS) 3 and SOCS4 expression in intestinal gut and causes disruption of inflammation cytokine production. These changes may affect the development of inflammatory bowel disease in heat-stressed pigs. However, the mechanisms are not well understood. Accordingly, in this study, we examined the roles of SOCS members in regulation of the nuclear factor (NF)-κB pathway and heat shock protein (HSP) 70-mediated cytokine induction in 293T human embryonic kidney cells and IPEC-J2 porcine small intestinal epithelial cells. Ectopic expression of HSP70 significantly modulated NF-κB activity (p ≤ .05). Moreover, co-expression of SOCS3 or SOCS4 with HSP70 reduced NF-κB activity, which was abolished by SOCS3 or SOCS4 knockdown with short hairpin RNA. Interestingly, MyD88-adaptor-like (Mal) protein was downregulated in cells expressing SOCS3 but not in cells expressing SOCS4. In addition, SOCS3 but not SOCS4 negatively regulated the activity of NF-κB induced by HSP70 overexpression via degradation of Mal. These findings may facilitate the development of novel SOCS3-based therapeutic strategies to control heat stress-related disorders in pigs.
Introduction
The risk of heat stress in pigs is increasing due to global warming and high stocking densities. Heat stress in pigs induces immunosuppression and cytokine disorders, thereby increasing susceptibility to diseases [Citation1,Citation2]. Heat-shock proteins (HSPs) act as chaperones and are involved in various cellular functions, such as protein folding, trafficking and membrane translocation [Citation3,Citation4]. Moreover, these proteins are induced in heat-stressed animals [Citation5]. For example, HSP70 is a critical immune modulator that induces cytokine production [Citation6,Citation7] and is upregulated under heat stress in pigs [Citation8]. However, the role of HSP70 in heat stress-induced immunosuppression in pigs is still unclear.
Cytokines play important roles in modulation of physiological systems. The suppressors of the cytokine signaling (SOCS) family is a family of important immune regulators in mammals [Citation9]. Eight family members, SOCS1–7 and CIS, have been identified, and every member has an amino-domain of variable length and divergent sequence, a central SH2 domain and a C-terminal 40-amino acid motif known as the SOCS box. SOCS proteins can be rapidly induced by various cytokines in a tissue-specific manner [Citation10,Citation11]. Additionally, these proteins participate in a negative-feedback loop modulating cytokine-induced signaling pathways [Citation12,Citation13]. Cytokines such as interleukin (IL)-6 and IL-7 induce the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling pathway, leading to expression of SOCS. Induced SOCS proteins bind directly via their SH2 domains to tyrosine-phosphorylated JAK or activated cytokine receptors, resulting in inhibition of cytokine signaling [Citation14].
SOCS members can also inhibit cytokine signaling by SOCS-box-mediated ubiquitination to induce targeted proteasomal degradation of signaling proteins [Citation15,Citation16]. SOCS1-deficient mice are highly sensitive to lipopolysaccharide (LPS)-induced shock and increased inflammatory cytokines [Citation17]. Moreover, SOCS1 inhibits LPS-induced nuclear factor (NF)-κB and STAT1 activation in macrophages [Citation17]. Similar to SOCS1, SOCS3 also rapidly increases in response to IL-6 and later downregulates IL-6 by preventing interactions among JAK, STAT3, and other signaling proteins [Citation18]. However, in chronic hypoxia of cardiocytes, SOCS3 regulates cell signaling crosstalk between NF-κB and phospho-STAT3 to maintain a balance between them for adaptation of myocardial cells to chronic hypoxia [Citation19]. In addition, SOCS3 exerts broader effects on immune responses by inhibiting signals induced by cytokines, such as type I and type II interferons and IL-12 [Citation20–22].
In previous reports, we showed that both SOCS3 and SOCS4 are upregulated in heat-stressed pigs in a time- and tissue-dependent manner [Citation23,Citation24]. However, the mechanisms mediating the interaction between SOCS and HSP70 are still unclear. In this study, we investigated the mechanisms of SOCS3 and SOCS4 regulation of HSP70-mediated NF-κB activity in 293T cells and IPEC-J2 cells.
Material and methods
Cell culture and heat shock
IPEC-J2 cells were seeded and incubated in 12-well tissue culture plates in culture medium (RPMI 1640; Gibco, Grand Island, NY) at a concentration of 2 × 105 cells/mL. One culture plate was incubated at 42 °C (95% air, 5% CO2, 100% humidity) for 1.5 h and then incubated at 37 °C for 1.5 h (heat-treated cells). Control cells were incubated at 37 °C (95% air, 5% CO2, 100% humidity) for 3 h without any treatment (control cells). Cells were collected at 0, 0.5, 1 and 1.5 h after heat shock for subsequent detection.
Construction of porcine SOCS3, SOCS4 and HSP70 expressing vectors
Porcine SOCS3, SOCS4 and HSP70 sequences containing the corresponding full-length open-reading frame (ORF) were ligated separately into the mammalian expression vector pcDNA3.1/V5-His-TOPO TA (Invitrogen, Shanghai, China). The mammalian expression vector pcDNA3.1/V5-His-TOPO TA was purchased from Invitrogen. The sequences of small interfering RNAs (siRNAs) were as follows: SOCS3, 5′-GCUCAAGCUGGUGCAUCACUACAUG-3′ (forward) and 5′-CAUGUAGUGAUGCACCAGCUUGAGC-3′ (reverse); SOCS4, 5′-UAACCAUCCUUUCUGUCAGCACUUC-3′ (forward) and 5′-GAAGUGCUGACAGAAAGGAUGGUUA-3′ (reverse). Preliminary experiments to evaluate knockdown efficiency showed that both siRNAs reduced expression by approximately 90%. Plasmid DNAs were prepared with an endotoxin-free Plasmid Maxi Kit (Qiagen, Valencia, CA). 293T cells and IPEC-J2 cells were transfected with plasmid DNAs using Lipofectamine-3000 (Invitrogen).
Detection of genes by quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR)
Total RNAs were isolated from cells using the RNAiso Plus kit (TaKaRa, Shuzo, Kyoto, Japan). RNAs were reverse transcribed to cDNAs using QUANTITECT (Qiagen). Gene detection was performed using a SYBR green polymerase mix (TaKaRa). The primers used for qRT-PCR were designed with Primer3 software (http://www.broad.mit.edu/cgi-in/primer/primer3-www.cgi). Primer pairs () were selected for their specificity based on dissociation curves. For assay validation, purified products were cloned into pMD19-T (TaKaRa) and sequenced to verify the correct target amplification. PCR products were amplified using a LightCycler 480 (95 °C, 15 s; 61 °C, 15 s; 72 °C, 15 s, 30 cycles). The efficiency of PCR amplification was found to be greater than 97% for each primer set using six serial dilutions of cloned products as templates. The data are presented as means ± standard deviations. Data normalization was carried out based on expression of the β-actin gene, and quantification was carried out using the delta Ct method.
Table 1. Primer sequences and amplicon characteristics.
Luciferase reporter assay
Cells were seeded in 96-well plates at 4 × 104 cells/well in complete Dulbecco’s modified Eagle’s medium. Cells were transfected with plasmid DNAs after 24 h using Lipofectamine-3000 (Invitrogen) according to the manufacturer’s instructions [Citation25]. Briefly, cells in each well were transfected with the following plasmids: 60 ng pNF-κB (which expresses firefly luciferase under the control of NF-κB; Invitrogen) and 0.2 ng PRL-YK (which expresses Renilla luciferase constitutively via a β-actin promoter and serves to normalize transfection variables; Invitrogen). Twenty-four hours post-transfection, cells were stimulated with ligands (tumor necrosis factor [TNF] α; PeproTech Company, Rocky Hill, NJ) for 6 h. The cells were washed with phosphate-buffered saline (PBS) and lysed in passive lysis buffer (Promega, Shanghai, China). Luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase values were normalized by dividing light output for firefly luciferase by light output for Renilla luciferase. The experiments were repeated three times.
Extraction of protein
Cells (2 × 106) were rinsed twice with cold PBS, and cytoplasmic proteins were extracted using M-TERTM mammalian protein extraction reagent containing a protease inhibitor mixture (Roche Molecular Biochemicals, Mannheim, Germany). Protein concentrations were measured using a BCA-200 protein assay kit (Pierce, Rockford, IL).
Western immunoblotting
Immunoblotting was performed as described previously [Citation26]. In brief, proteins were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electro-transferred to nitrocellulose membranes. After blocking in 25 mM Tris-HCl (pH 7.5), 1.25 mM NaCl and 0.1% Tween 20 (TBST) with 5% fat-free milk, blots were incubated with primary antibodies for 2 h and then with horseradish-peroxidase-conjugated secondary antibodies for 1 h. Rabbit anti-human IκBα polyclonal antibodies and rabbit anti-human phospho-IκBα polyclonal antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and rabbit anti-pig Mal monoclonal antibodies and rabbit-anti-β-actin IgG were purchased from Proteintech (vendor location). Peroxidase-conjugated Affinipure Goat anti-Rabbit IgG antigen/antibody complexes on membranes were detected using a SuperSignal Ultra chemoluminescence kit (Thermo Scientific, Waltham, MA). Protein levels were quantified using densitometry analysis and are expressed in OD units of target protein relative to β-actin.
Statistical analysis
Data are presented as the means ± standard errors. Statistical significance was determined by analysis of variance (ANOVA), followed by Student’s t-tests. Differences with p values of less than .05 were considered significant.
Results
Effects of heat stress on HSP70 expression and NF-κB activity
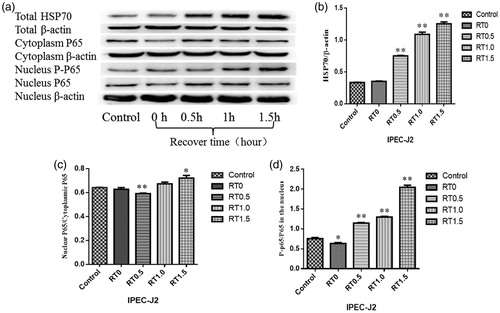
In order to elucidate the effects of heat stress on HSP70 expression and NF-κB activity, we detected the expression levels of HSP70, cytosolic p65 and nuclear p65 in IPEC-J2 cells challenged by heat stress for 1.5 h and then allowed to recover for 1.5 h. shows changes in HSP70, cytosolic p65 and nuclear p65 in IPEC-J2 cells under conditions of heat stress. There were no significant differences in HSP70 levels between control and heat-stressed IPEC-J2 cells at 0 h after treatment; however, at 0.5, 1 and 1.5 h, HSP70 levels were higher in heat-stressed cells than that in control cells (p < .01). Moreover, the ratio of nuclear phosphorylated p65 to total p65 significantly increased at 0.5, 1 and 1.5 h (p < .01). The results showed that the level of pNF-kappa-B increased in IPEC-J2 cells under heat stress for 1.5 h and recover for 1.5 h and that HSP 70 was up-regulated.
Figure 1. Effects of heat stress on HSP70 expression and NF-κB activity. The levels of HSP70, cytosolic and nuclear p65, and phospho-p65 (a) in IPEC-J2 cells challenged by heat stress for 1.5 h followed by recovery for 1.5 h. Changes in HSP70 expression (b). Ratios of nuclear and cytoplasmic p65 heat stress (c). Ratios of nuclear phospho-p65 to p65 after heat stress for (d). *p ≤ .05, **p ≤ .01.
Ectopic expression of porcine SOCS3, SOCS4 and HSP70
To construct expression vectors for SOCS3, SOCS4 and HSP70, we used a PCR technique to isolate cDNA fragments of these genes () from porcine PBMCs and inserted cDNA fragments into the PMD18-T vector (TaKaRa). Porcine SOCS3 (pSOCS3) and SOCS4 (pSOCS4) cDNA gene fragments were then ligated into the pcDNA3.1/V5-His-TOPO vector with a His-tag at the C-terminus, yielding pcDNA3.1/V5-SOC3-His and -SOC4-His.
Figure 2. Construction of an SOCS3, SOCS4 and HSP70 ectopic expression system and cotransfection in vitro. The SDS-PAGE: PCR product of the SOCS3 plasmid (a), PCR product of pcDNA 3.1/V5-His-SOCS3 (b), PCR product of the SOCS4 plasmid (c) and PCR product of pcDNA 3.1/V5-His-SOCS4 (d). Western blot analysis of SOCS3 (e), HSP70 (f) and SOCS4 proteins (g) in 293 T cells that transfected by their ectopic expression vectors, respectively. The expressing of HSP70 in supernatant of cells that co-transfected by HSP70 + pNF-κB-Luc + pRL-YK also was detected (h). qRT-PCR analysis of HSP70 and SOCS3 mRNA levels in 293T cells (i) and IPEC-J2 cells (j) transfected with empty, HSP70 + pNF-κB-Luc + pRL-YK, SOCS3 + pNF-κB-Luc + pRL-YK or HSP70 + SOCS3 + pNF-κB-Luc + pRL-YK vectors. HSP70 and SOCS4 expression in 293T cells (k) and IPEC-J2 cells (l) transfected by empty, HSP70 + pNF-κB-Luc + pRL-YK, SOCS4 + pNF-κB-Luc + pRL-YK or HSP70 + SOCS4 + pNF-κB-Luc + pRL-YK vectors. Empty refers to pcDNA 3.1/V5-His vector that was not linked to a gene. *p ≤ .05, **p ≤ .01. Lanes 1 and 2 in (b) show PCR products of the SOCS3-ORF and pcDNA 3.1/V5-His-SOCS3 vectors; lanes 1 and 2 in (d) show the PCR products of the SOCS4-ORF and pcDNA 3.1/V5-His-SOCS4 vectors.
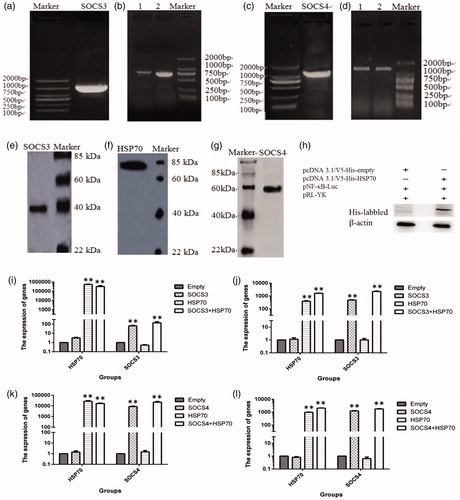
To establish a cotransfection model system, DNA plasmids of pSOCS3, pSOCS4 and pHSP70 and the NF-κB reporter system (pNF-κB and pRL-YK) were cotransfected into 293T cells and IPEC-J2 cells. We used qRT-PCR to measure the expression of the HSP70, SOCS3 and SOCS4 genes. Increased transcripts of HSP70 plus SOCS3 (p < .01, ) and HSP70 with SOC4 (p < .01, ) were detected in both cotransfected cell lines. Western blotting was used to verify the expression of pSOCS3, pSOCS4 and pHSP70, and these proteins were obviously expressed (). The protein sequences of pSOCS3, pSOCS4 and pHSP70 were verified by sequencing analysis. Extracellular HSP70 in IPEC-J2 cells transfected with pHSP70 and the NF-κB reporter system was also detected ().
HSP70 overexpression upregulated the activity of NF-κB in cultured cells
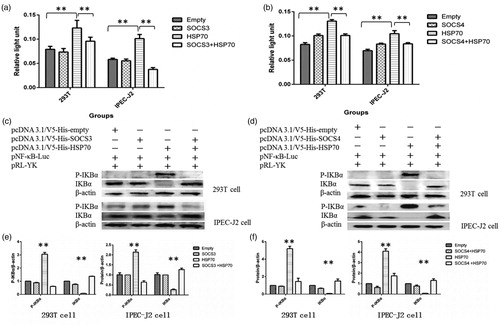
To detect effects of HSP70 on activity of NF-κB, plasmids encoding pHSP70 and the NF-κB reporter system (pNF-κB and pRL-YK) were transiently cotransfected into 293T cells and IPEC-J2 cells. Stimulation of cells with TNF-α (50 ng/mL) resulted in increased activity of NF-κB in treated cells but not in control cells (). Immunoblotting showed that the expression of IKBα (an inhibitor of NF-κB) was lower in pHSP70-transfected cells () and that the levels of phosphorylated IKBα (phospho-IKBα) were higher than those in the controls (). These results indicated that the activity of NF-κB was induced in HSP70-overexpressing cells.
Figure 3. SOCSs decreased the activity of NF-κB induced by HSP70 overexpression. Cells were cotransfected with the NF-κB reporter system (pNF-κB and pRL-YK) and SOCS3 + HSP70 (a) or SOCS4 + HSP70 (b) and then exposed to TNF-α for 6 h. Finally, activation of NF-κB was measured using dual luciferase reporter assays. The levels of IKBα and phospho-IKBα were detected by western blotting in 293T cells and IPEC-J2 cells transfected with SOCS3 + HSP70 (c, e) and SOCS4 + HSP70 (d, f). Data in (e) and (f) are from one representative experiment with three technical replicates (means ± standard deviations) and were statistically analyzed by one-way ANOVA. **p ≤ .01. All experiments were repeated three times.
SOCS3 and SOCS4 negatively regulated HSP70-induced NF-κB activity
To elucidate the interactions of SOCS, HSP70 and NF-κB, the pSOCS3 or pSOCS4, pHSP70 and NF-κB reporter genes (pNF-κB and pRL-YK) were cotransfected into 293T cells and IPEC-J2 cells. NF-κB reporter luciferase assays showed that ectopic expression of either SOCS3 or SOCS4 resulted in suppression of pHSP70-induced NF-κB gene expression in both 293T and IPEC-J2 cells (p < .01, ). Expression of either SOCS3 or SOCS4 also reduced the levels of HSP70-induced phospho-IKBα (p < .01; ). These results indicated that both SOCS3 and SOCS4 negatively regulated the activity of NF-κB induced by HSP70 overexpression in 293T and IPEC-J2 cells.
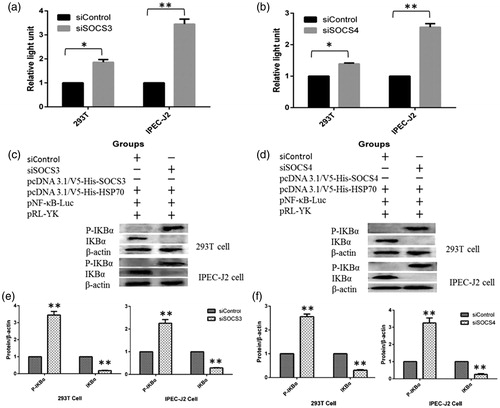
Knockdown of SOCS3 or SOCS4 upregulated NF-κB in HSP70-expressing cells
We also used specific short hairpin RNAs (shRNAs) to knockdown SOCS3 and SOCS4. After cotransfection, NF-κB activity was increased with reduced expression of SOCS3 or SOCS4 in 293T and IPEC-J2 cells (). Protein levels of IKBα were also reduced by knocking down SOCS3 () or SOCS4 (); however, phospho-IKBα levels were increased significantly compared with that in the controls. The results further indicated that SOCS3 and SOCS4 were negative regulators of NF-κB in HSP70-expressing cells.
Figure 4. SOCSs knockdown abolished NF-κB activity in HSP70-expressing cells. Cells were transfected with the NF-κB reporter system (pNF-κB and pRL-YK) and siSOCS3 + SOCS3 + HSP70 (a) or siSOCS4 + SOCS4 + HSP70 (b), and the activation of NF-κB was measured using dual luciferase reporter assays. The levels of IKBα and phospho-IKBα were detected by western blotting in 293T cells and IPEC-J2 cells transfected with siSOCS3 + SOCS3 + HSP70 (c, e) and siSOCS4 + SOCS4 + HSP70 (d, f). Data in (e) and (f) are from one representative experiment with three technical replicates (means ± standard deviations) and were statistically analyzed by one-way ANOVA. *p ≤ .05, **p ≤ .01. All experiments were repeated three times.
SOCS3, but not SOCS4, decreased the expression of Mal protein induced by HSP70
To further elucidate the mechanisms through which pSOCS3 and pSOCS4 regulate HSP70-induced NF-κB activity, we studied the Mal protein, an upstream modulator of the NF-κB pathway. Ectopic expression of HSP70 resulted in elevation of Mal protein in both 293T and IPEC-J2 cells (). Co-expression of SOCS3 with HSP70 blocked Mal elevation (). Knocking down SOCS3 with shRNA restored HSP70-induced Mal elevation (). Interestingly, the same dynamic changes were not detected in cells cotransfected with shRNA targeting pSOCS4, pHSP70 and pSOCS4 (). The results showed that SOCS3, but not SOCS4, controlled the activity of NF-κB induced by HSP70 via degradation of Mal protein (.
Figure 5. SOCS3, but not SOCS4, decreased the expression of MyD88-adapter-like (Mal) protein induced by HSP70. Changes in the expression of Mal protein in HSP70-overexpressing cells and cells with SOCS3 cotransfection (a, b). Changes in Mal protein expression following cotransfection with SOCS4 and HSP70 (c–e). Data in (b) and (e) are from one representative experiment with three technical replicates (means ± standard deviations) and were analyzed by one-way ANOVA. **p ≤ .01. All experiments were repeated three times.
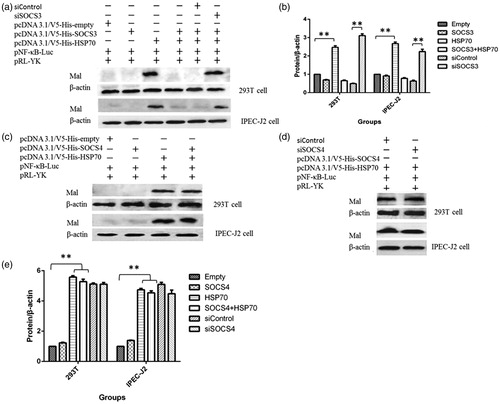
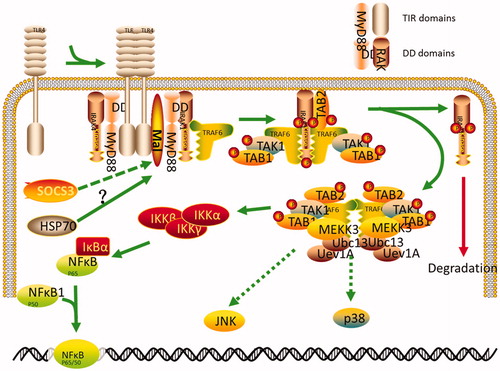
Figure 6. The putative signaling pathway of SOCS3 in HSP70-overexpressing cells. HSP70 released from HSP70-transfected cells may interact with TLR2 or TLR4 in these cells and lead to activation of NF-κB. This pathway can then be disrupted by SOCS3 or SOCS4 cotransfection. SOCS3, but not SOCS4, negatively regulates the activity of NF-κB via degradation of Mal protein in 293T cells and IPEC-J2 cells.
Discussion
The NF-κB pathway is a pro-inflammatory pathway owing to the role of NF-κB in the regulation of pro-inflammatory genes, including cytokines, chemokines and adhesion molecules [Citation27]. The activity of NF-κB is inhibited by heat stress in RAW264.7 macrophoages [Citation28]. However, in dendritic cells, the activity of NF-κB is enhanced under heat stress (41 °C) and oxidative stress [Citation29]. Interestingly, in the human monocytic cell line U937, overexpression of HSP70 activates NF-κB and activator protein-1, which stimulates matrix metalloproteinase-9 [Citation30]. In the soleus muscle of rats, HSP70 overexpression abolishes the transcriptional activities of NF-κB, thereby preventing atrophy of disused muscle fiber [Citation31]. In this study, the activity of NF-κB was significantly upregulated in HSP70-expressing cells, indicating that changes in NF-κB activity in HSP70-overexpressing cells may depend on the cell type.
Several reports have shown that SOCS3 regulates Toll-like receptor (TLR) signaling. LPS treatment strongly induces SOCS3 expression in macrophages and hepatocytes [Citation32]. In addition, similar to SOCS1, SOCS3 inhibits TLR-mediated responses [Citation33]. However, the role of SOCS3 in TLR signaling may be complex; indeed, conflicting reports have demonstrated that SOCS3 has little or sometimes an enhancing effect on the TLR response [Citation34,Citation35]. SOCS3 has also been shown to inhibit type I interferon signaling via an NF-κB-dependent pathway [Citation22]. In this study, SOCS3/4 overexpression negatively regulated the activity of NF-κB. We speculate that extracellularly released HSP70 may interact with TLR2 or TLR4 in these cells and lead to activation of NF-κB. Moreover, this pathway could be disrupted by SOCS3/4 cotransfection. However, more studies are required to confirm these findings.
The TLR adaptor molecule Mal/TIRAP is a target of SOCS1 [Citation36]. TLR stimulation induces Btk-dependent tyrosine-phosphorylation of Mal and generates a binding site for the SH2 domain of SOCS1. Subsequent interaction between SOCS1 and Mal induces ubiquitination and proteasomal degradation of Mal, resulting in elimination of TLR/Mal-dependent NF-κB activation [Citation36]. Our results showed that SOCS3 also induced the degradation of Mal and inhibition of NF-κB in HSP70-overexpressing cells, probably because SOCS3 and SOCS1 possess a kinase-inhibitory region domain, which is critical for inhibition of kinase activity [Citation37].
Although extensive studies on the role of SOCS3 have been reported, the targets and pathways regulated by SOCS4 are still unclear. In vitro studies have suggested that SOCS4 is involved in regulating epidermal growth factor signaling [Citation38], may be regulated by parasitic infections [Citation39], is a critical regulator of antiviral immunity [Citation40], is linked to better outcomes in patients with cancer [Citation41] and even regulates pre-granulosa cells during folliculogenesis [Citation42]. Similar to SOCS3, SOCS4 also negatively regulates NF-κB activity induced by HSP70 overexpression; however, we did not observe degradation of the Mal protein in SOCS4 and HSP70 cotransfected cells. This may be due to the different structures of SOCS3 and SOCS4. The N-terminal region of SOCS4 is extended compared with that in SOCS3; such an extension may assist in providing a supplementary level of negative regulation not observed in the other SOCS proteins [Citation43,Citation44].
Overall, our findings suggested that SOCS3 but not SOCS4 was able to negatively regulate HSP70-induced NF-κB via degradation of Mal in porcine cells. These results may have important insights into the development of therapeutic strategies for the control of heat stress-related disorders in animals.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Ju X-H, Yong Y-H, Xu H-J, et al. Impacts of heat stress on baseline immune measures and a subset of T cells in Bama miniature pigs. Livestock Sci. 2011;135:289–292.
- Ju X-H, Xu H-J, Yong Y-H, et al. Heat stress upregulation of Toll-like receptors 2/4 and acute inflammatory cytokines in peripheral blood mononuclear cell (PBMC) of Bama miniature pigs: an in vivo and in vitro study. Animal. 2014;8:1462–1468.
- Evans CG, Chang L, Gestwicki JE. Heat shock protein 70 (hsp70) as an emerging drug target. J Med Chem. 2010;53:4585–4602.
- Wisén S, Bertelsen EB, Thompson AD, et al. Binding of a small molecule at a protein–protein interface regulates the chaperone activity of hsp70-hsp40. ACS Chem Biol. 2010;5:611–622.
- Banerjee MS, Chakraborty PK, Dey RS, et al. Heat stress upregulates chaperone heat shock protein 70 and antioxidant manganese superoxide dismutase through reactive oxygen species (ROS), p38MAPK, and Akt. Cell Stress Chaperones. 2009;14:579–589.
- Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305.
- Asea A, Kraeft S-K, Kurt-Jones EA, et al. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435–442.
- Zhong X, Wang T, Zhang X, et al. Heat shock protein 70 is upregulated in the intestine of intrauterine growth retardation piglets. Cell Stress Chaperones. 2010;15:335–342.
- Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001;19:378–387.
- Endo TA, Masuhara M, Yokouchi M, et al. A new protein containing an SH2 domain that inhibits JAK kinases. Nature. 1997;387:921–924.
- Hilton DJ, Richardson RT, Alexander WS, et al. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc Nat Acad Sci. 1998;95:114–119.
- Starr R, Willson TA, Viney EM, et al. A family of cytokine-inducible inhibitors of signalling. Nature. 1997;387:917–921.
- Naka T, Narazaki M, Hirata M, et al. Structure and function of a new STAT-induced STAT inhibitor . Nature. 1997;387:924–929.
- Krebs DL, Hilton DJ. SOCS: physiological suppressors of cytokine signaling. J Cell Sci. 2000;113:2813–2819.
- Zhang J-G, Farley A, Nicholson SE, et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Nat Acad Sci. 1999;96:2071–2076.
- Kamizono S, Hanada T, Yasukawa H, et al. The SOCS box of SOCS-1 accelerates ubiquitin-dependent proteolysis of TEL-JAK2. J Biol Chem. 2001;276:12530–12538.
- Nakagawa R, Naka T, Tsutsui H, et al. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17:677–687.
- Xiang S, Dong N-G, Liu J-P, et al. Inhibitory effects of suppressor of cytokine signaling 3 on inflammatory cytokine expression and migration and proliferation of IL-6/IFN-纬-induced vascular smooth muscle cells. J Huazhong Univ Sci Technol [Med Sci]. 2013;33:615–622.
- Gu Q, Kong Y, Yu Z-B, et al. Hypoxia-induced SOCS3 is limiting STAT3 phosphorylation and NF-κB activation in congenital heart disease . Biochimie. 2011;93:909–920.
- Qin H, Niyongere SA, Lee SJ, et al. Expression and functional significance of SOCS-1 and SOCS-3 in astrocytes. J Immunol. 2008;181:3167–3176.
- Qin H, Wilson CA, Roberts KL, et al. IL-10 inhibits lipopolysaccharide-induced CD40 gene expression through induction of suppressor of cytokine signaling-3. J Immunol. 2006;177:7761–7771.
- Pauli E-K, Schmolke M, Wolff T, et al. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. 2008;4:e1000196.
- Wang P, Ju X-H, Yong Y-H, et al. Cloning and bioinformatics analysis of porcine SOCS3 cDNA and its expression pattern during heat stress. Acta Veterinaria Zootechnica Sin. 2014;45:1044–1052.
- Wang P, Ju X-H, Yong Y-H, et al. Cloning and bioinformatics analysis of porcine SOCS4 cDNA and its expression pattern during heat stress. Acta Veterinaria Zootechnica Sin. 2015;46:323–331.
- Zhu J, Lai K, Brownile R, et al. Porcine TLR8 and TLR7 are both activated by a selective TLR7 ligand, imiquimod. Mol Immunol. 2008;45:3238–3243.
- An H, Xu H, Yu Y, et al. Up-regulation of TLR9 gene expression by LPS in mouse macrophages via activation of NF-κB, ERK and p38 MAPK signal pathways. Immunol Lett. 2002;81:165–169.
- Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651.
- Li C-L, Wang X-Y, Shao J, et al. Heat shock inhibits IL-12 p40 expression through NF-kappa B signalling pathway in murine macrophages. Cytokine. 2001;16:153–159.
- Wang Y, Seidl T, Whittall T, et al. Stress-activated dendritic cells interact with CD4+ T cells to elicit homeostatic memory . Eur J Immunol. 2010;40:1628–1638.
- Lee K-J, Kim YM, Kim DY, et al. Release of heat shock protein 70 (Hsp70) and the effects of extracellular Hsp70 on matric metalloproteinase-9 expression in human monocytic U937 cells. Exp Mol Med. 2006;38:364–374.
- Senf SM, Dodd SL, McClung JM, et al. Hsp70 overexpression inhibits NF-κB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J. 2008;22:3836–3845.
- Yang X-P, Schaper F, Teubner A, et al. Interleukin-6 plays a crucial role in the hepatic expression of SOCS3 during acute inflammatory processes in vivo. J Hepatol. 2005;43:704–710.
- Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465.
- Prele CM, Woodward EA, Bisley J, et al. SOCS1 regulates the IFN but not NFkappaB pathway in TLR-stimulated human monocytes and macrophages . J Immunol. 2008;181:8018–8026.
- Liu X, Zhang Y, Yu Y, et al. SOCS3 promotes TLR4 response in macrophages by feedback inhibiting TGF-beta1/Smad3 signaling . Mol Immunol. 2008;45:1405–1413.
- Mansell A, Smith R, Doyle SL, et al. Suppressor of cytokine signaling 1 negatively regulates Toll-like receptor signaling by mediating Mal degradation. Nat Immunol. 2006;7:148–155.
- Fujimoto M, Naka T. Regulation of cytokine signaling by SOCS family molecules. Trends Immunol. 2003;24:659–666.
- Kario E, Marmor MD, Adamsky K, et al. Suppressors of cytokine signaling 4 and 5 regulate epidermal growth factor receptor signaling. J Biol Chem. 2005;280:7038–7048.
- Hu G, Zhou R, Liu J, et al. MicroRNA-98 and let-7 regulate expression of suppressor of cytokine signaling 4 in biliary epithelial cells in response to Cryptosporidium parvum infection. J Infect Dis. 2010;202:125–135.
- Kedzierski L, Linossi EM, Kolesnik TB, et al. Suppressor of cytokine signaling 4 (SOCS4) protects against severe cytokine storm and enhances viral clearance during influenza infection. PLoS Pathog. 2014;10:e1004134.
- Sasi W, Jiang WG, Sharma A, et al. Higher expression levels of SOCS 1, 3, 4, 7 are associated with earlier tumour stage and better clinical outcome in human breast cancer. BMC Cancer. 2010;10:1.
- Sutherland JM, Keightley RA, Nixon B, et al. Suppressor of cytokine signaling 4 (SOCS4): moderator of ovarian primordial follicle activation. J Cell Physiol. 2012;227:1188–1198.
- Feng ZP, Chandrashekaran IR, Low A, et al. The N-terminal domains of SOCS proteins: a conserved region in the disordered N-termini of SOCS4 and 5. Proteins. 2012;80:946–957.
- Flowers LO. SOCS negative regulation of the JAK-STAT pathway. Int J Biosci. 2012;2:13–23.