
Abstract
Purpose: Radiofrequency ablation (RFA) is widely accepted as a curative treatment for small hepatocellular carcinoma (HCC). However, insufficient RFA (IRFA) can lead to rapid local recurrence. The underlying mechanisms remain poorly understood. This study aimed to elucidate the role and regulatory mechanisms of autophagy in the recurrence of HCC after IRFA.
Materials and methods: SMMC7721 and Huh7 cells were exposed to sublethal heat stress to stimulate the transition zone of IRFA treatment. The levels of autophagy were measured by western blot, immunofluorescence and transmission electron microscopy. Functional assays, such as CCK-8, EdU incorporation and flow cytometry, were performed to determine the role of heat-induced autophagy. The involved signaling pathways were explored by western blot. Finally, the antitumor effects of chloroquine (CQ) on heat-treated HCC cells were evaluated via an in vivo xenograft tumor model.
Results: Sublethal heat stress induced autophagy in a temperature- and time-dependent manner in HCC cells. Furthermore, the inhibition of autophagy by CQ or siRNA targeting the autophagy-related genes Beclin-1 and Atg5 enhanced heat-induced apoptosis. The combination of CQ and heat treatment significantly suppressed tumor growth both in vitro and in vivo. Mechanistically, we reported for the first time that the ATP-AMPK-mTOR signaling pathway was involved in heat-induced autophagy.
Conclusions: Heat stress induced protective autophagy against heat-induced apoptosis in HCC via the ATP-AMPK-mTOR axis, suggesting that targeting autophagy may be a promising strategy for improving the efficacy of RFA treatment for HCC.
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common malignancy globally and the third most common cause of cancer-related death [Citation1–3]. Radiofrequency ablation (RFA), a type of hyperthermia treatment, has been accepted as a curative therapy alternative to surgical resection and liver transplantation for small HCC due to such advantages as minimal invasiveness, safety, repeatability and shorter hospitalization time [Citation4]. However, one of the major problems with RFA is that insufficient RFA (IRFA) may lead to residual tumor progression and rapid local recurrence. Although IRFA can cause the malignant transformation of HCC via epithelial-mesenchymal transition (EMT) or the hypoxia-inducible factor-1α (HIF-1α)/VEGFA pathway [Citation5–7], the specific molecular mechanisms remain poorly understood.
Autophagy is an evolutionarily conserved process by which damaged or excessive organelles and cytoplasmic proteins are degraded by the action of lysosomes [Citation8]. Autophagy can be activated by multiple signaling pathways in response to a number of cellular stresses, including starvation, hypoxia, oxidation, radiation and chemotherapy [Citation9]. Autophagy appears to be a double-edged sword in tumor development; whether autophagy promotes cell survival or induces cell death depends on the cell type and environmental stress [Citation10,Citation11]. Previous studies showed that sublethal heat stress induces autophagy in several human cancer cell lines, such as HeLa and A549 cells [Citation12]. Recent studies reported that IRFA promotes the progression of residual HCC via autophagy [Citation13,Citation14]. These findings strongly support a correlation between autophagy and HCC progression after IRFA. However, the precise regulatory mechanisms and signaling pathways underlying IRFA-induced autophagy remain largely unknown.
In this study, we focused on whether sublethal heat stress can induce autophagy in HCC cells. We aimed to decipher the role of autophagy in heat-treated HCC cells and explore the precise regulatory mechanisms and signaling pathways underlying heat-induced autophagy. Our findings may provide a new basis for understanding heat-induced autophagy in residual tumor progression and rapid local recurrence of HCC after IRFA treatment. This study indicated that targeting autophagy may be a promising therapeutic strategy for improving the efficacy of RFA treatment for HCC.
Materials and methods
Cell culture and reagents
The human HCC cell lines SMMC7721 and Huh7 were obtained from the Institute of Biochemistry and Cell Biology, Chinese Academy of Science (Shanghai, China). Cells were cultured in RPMI 1640 medium (Gibco, Grand Island, NY) containing 10% fetal bovine serum (FBS) (Zeta, Menlo Park, CA), 100 U/mL penicillin and 100 mg/mL streptomycin (Invitrogen, Carlsbad, CA) at 37 °C and 5% CO2. To measure autophagy and autophagy flux, SMMC7721 and Huh7 cells were transfected with mRFP-GFP-LC3 adenovirus (HanBio, Shanghai, China) as described previously [Citation15]. The following reagents were used in this study: chloroquine (CQ) (C6628) (Sigma-Aldrich, St. Louis, MO), Lipofectamine 3000 (Invitrogen, Carlsbad, CA), 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) (S1802) and compound C (S7840) (Selleck, Houston, TX).
Heat treatment
Heat treatment was carried out by sealing the mouths of T25 flasks or the tops of culture plates with parafilm and submerging the flasks or plates in a water bath at 43, 45 or 47 °C for the indicated duration. After heat treatment, cells were cultured at 37 °C for recovery. Cells incubated at 37 °C only served as the control group.
Western blot
Cells were collected and lysed in RIPA buffer (Beyotime Biotechnology, Shanghai, China). Subsequently, the proteins were separated by 8–12% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Burlington, MA), which were blocked with 5% nonfat milk and incubated with primary and secondary antibodies. Afterwards, the protein bands were visualized with an ECL detection kit (Millipore, Burlington, MA) and analyzed using Quality One 1-D analysis software (Bio-Rad, Hercules, CA). The following antibodies were used: LC3B (L7543, Sigma-Aldrich, St. Louis, MO), SQSTM1/p62 (#5114S), Beclin-1 (#3495S), Atg5 (#12994S), Atg7 (#8558S), cleaved PARP (#5625S), cleaved Caspase-3 (#9664S), phospho-AMPKα (Thr172) (#2535S), AMPKα (#2532S), phospho-mTOR (Ser172) (#2971S), mTOR (#2972S), phospho-p70S6Kinase (p70S6K) (Thr389) (#9234S), p70S6Kinase (#9202S), phospho-4E-BP1 (Ser65) (#9451S), 4E-BP1 (#9644S) (Cell Signaling Technology, Danvers, MA); and β-actin (20536–1-AP), horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody (SA00001-2) (Proteintech, Rosemont, IL).
Confocal microscopy
Cells were transfected with mRFP-GFP-LC3 adenovirus, followed by heat treatment. Then, cells were fixed with 4% paraformaldehyde for 15 min and subjected to confocal microscopy (Zeiss, Oberkochen, Germany). Levels of autophagy and autophagy flux were determined by evaluating the numbers of GFP and mRFP puncta per cell.
Transmission electron microscopy
Transmission electron microscopy was used to identify and analyze the ultrastructures of autophagosomes/autolysosomes as described previously [Citation16,Citation17].
Cell viability and EdU incorporation assay
Cells were seeded into 96-well plates at a density of 1 × 104 cells per well and cultured overnight for attachment. Then, cells were exposed to CQ, heat treatment or both in combination. After recovery at 37 °C for another 24 h, cell viability was evaluated using a Cell Counting Kit-8 (CCK-8) (Dojindo, Tokyo, Japan). Cell proliferation was assessed by a Cell-Light™ EdU Apollo®488 In Vitro Imaging Kit (RiboBio, Guangzhou, China) according to the manufacturer’s instructions.
Flow cytometry assay
A FITC Annexin V/Dead Cell Apoptosis Kit (Invitrogen, Carlsbad, CA) was used to detect the apoptosis of cells exposed to different treatments. Annexin V-positive cells represented apoptotic cells and were quantified as previously described [Citation18].
RNA interference
Small interfering RNAs (siRNAs) targeting Beclin-1 or Atg5 were purchased from Sangon (Shanghai, China) and transfected into cells using Lipofectamine 3000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s guidelines. The siRNA sequences were as follows: negative control (NC) siRNA: 5′-UUCUCCGAACGUGUCACGUTT-3′, 3′-ACGUGACACGUUCGGAGAATT-5′; Beclin-1 siRNA 1: 5′-GGUCUAAGACGUCCAACAATT-3′, 3′-UUGUUGGACGUCUUAGACCTT-5′; Beclin-1 siRNA 2: 5′-GGAGCCAUUUAUUGAAACUTT-3′, 3′-AGUUUCAAUAAAUGGCUCCTT-5′; Beclin-1 siRNA 3: 5′-GCUGCCGUUAUACUGUUCUTT-3′, 3′-AGAACAGUAUAACGGCAGCTT-5′; and Atg5 siRNA: 5′-GCUUCGAGAUGUGUGGUUUTT-3′, 3′-GCUUCGAGAUGUGUGGUUUTT-5′. Atg5 siRNA was chosen according to a previous study [Citation19].
Measurement of cellular ATP
Cells were seeded into 96-well plates at a density of 1 × 104 cells per well and exposed to heat treatment. After recovery for another 24 h, ATP content was measured using the ATPlite Luminescence Assay Kit (#6016943) (PerkinElmer, Akron, OH) as described in our previous study [Citation20]. Relative ATP levels were normalized to the control.
In vivo xenograft tumor assay
Five-week-old male BALB/c nude mice were obtained from Bioscience (Beijing, China) and maintained in the laboratory animal center of the Army Medical University (Chongqing, China). Animal studies were approved by the Institutional Animal Care and Use Committee of Southwest Hospital (Chongqing, China). SMMC7721 cells were pretreated at 47 °C for 30 min or maintained at 37 °C. After recovery at 37 °C for 24 h, cells (5 × 106 cells in 100 μL of serum-free RPMI 1640 medium) were injected into the upper right flank of 24 nude mice. Nude mice were randomly separated into four groups (n = 6 mice per group): vehicle, CQ, heat treatment, and a combination of CQ and heat treatment. Starting on the second day, mice in vehicle and heat treatment groups received i.p. injections of 100 µl of PBS thrice weekly, while mice in CQ and combination treatment groups received i.p. injections of 60 mg/kg CQ in 100 µl of PBS thrice weekly. Tumor size was measured every 3 d for 30 d, and the results were plotted. After 30 d, tumors were harvested, photographed, weighed and then fixed in 4% paraformaldehyde for immunohistochemistry (IHC) staining.
IHC staining
Xenograft tumors were harvested and then fixed in 4% paraformaldehyde for 48 h. Then, the tumor tissues were embedded and sliced at a thickness of 5 μm. Subsequently, tumor sections were immunostained with a Histostain-Plus Kit (Zhongshan Biotechnologies, Beijing, China) according to the manufacturer’s instructions. Finally, the sections were photographed, and the intensity of LC3, p62, Ki-67 and cleaved Caspase-3 staining was quantified with Image-Pro Plus software (Media Cybernetics, Rockville, MD). The following primary antibodies were used: LC3A/B (#12741S), cleaved Caspase-3 (#9664S) (Cell Signaling Technology, Danvers, MA); and SQSTM1/p62 (18420–1-AP), Ki-67 (27309–1-AP) (Proteintech, Rosemont, IL).
Statistical analysis
Statistical analysis was performed using the GraphPad Prism version 5.0 (GraphPad Software, La Jolla, CA). Comparisons between two groups were performed by using Student’s t test. Multiple comparisons were performed by using ANOVA. Categorical data were analyzed using the chi-square test or Fisher’s exact test. A value of p < .05 was considered statistically significant.
Results
Sublethal heat stress induces autophagy in HCC cells
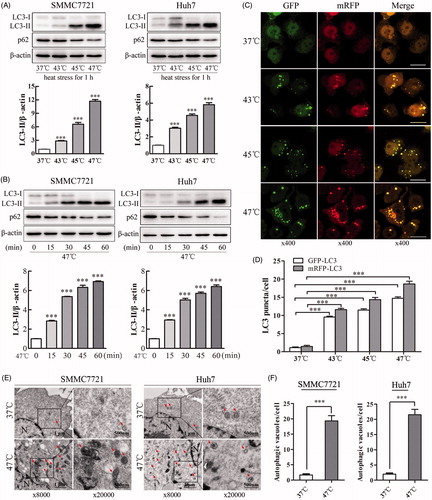
Sublethal heat stress induces autophagy in several types of cancer cells, such as HeLa and A549 cells [Citation12]. However, whether this phenomenon occurs in HCC cells remains unknown. The conversion of cytosolic LC3-I to LC3-II, which binds autophagic vacuoles, is the primary hallmark of autophagy. In addition, the protein p62 (also known as SQSTM1) has been reported to interact with the autophagic effector protein LC3 and to be degraded through an autophagy-lysosome pathway [Citation21]. To determine whether sublethal heat stress can induce autophagy in HCC cells, we exposed SMMC7721 and Huh7 cells to sublethal heat stress and then performed western blot analysis. Heat treatment increased LC3-II levels and decreased p62 levels in a temperature-dependent manner (). Consistently, heat treatment at 47 °C for various durations (0, 15, 30, 45 or 60 min) also increased LC3-II levels and decreased p62 levels in a time-dependent manner (). Furthermore, we utilized a tandem mRFP-GFP-LC3 adenovirus construct to confirm autophagy induction by the formation of GFP-LC3 and mRFP-LC3 puncta, which indicate autophagosome formation [Citation15]. As shown in and Supplemental Figure S1(A,B), heat stress induced the accumulation of GFP-LC3 and mRFP-LC3 puncta in the perinuclear region and cytoplasm in SMMC7721 and Huh7 cells in a temperature-dependent manner, suggesting that heat stress induces autophagy. As shown in Supplemental Figure S1(C), in contrast to the control cells, heat-treated SMMC7721 and Huh7 cells contained numerous microscopic vacuoles. To confirm these findings, typical double-membraned vacuolar structures with the morphological features of autophagosomes in heat-treated SMMC7721 and Huh7 cells were analyzed using a transmission electron microscope (). The number of autophagic vacuoles per cell was dramatically increased from less than 3 to 20 after heat treatment (). Taken together, these results indicate that sublethal heat stress induces autophagy in HCC cells.
Figure 1. Sublethal heat stress induces autophagy in HCC cells. (A, B) SMMC7721 and Huh7 cells were exposed to 37, 43, 45 or 47 °C for 60 min, followed by recovery at 37 °C for 8 h (A), or to 47 °C for 0, 15, 30, 45 or 60 min, followed by recovery at 37 °C for 24 h (B). The expression levels of LC3-II and p62 were analyzed by western blot. LC3-II levels were quantified and normalized to those of β-actin. (C, D) Representative images and quantification results of early autophagosomes in SMMC7721 cells transiently transfected with mRFP-GFP-LC3 adenovirus after exposure to 37, 43, 45 or 47 °C for 60 min and recovery at 37 °C for 8 h. The average numbers of GFP-LC3 and mRFP-LC3 puncta per cell were counted in 6 random fields with at least 100 cells in each group. Scale bar, 20 μm. (E, F) Transmission electron microscopy showed an increased number of autophagic vacuoles in SMMC7721 and Huh7 cells after exposure to 47 °C for 30 min. Typical autophagosomes (arrows); N, nucleus; Scale bar, 2 μm. Each experiment was repeated three times, and representative images are shown. Error bars represent the SEM. ***p < .001.
Sublethal heat stress activates autophagic flux in HCC cells
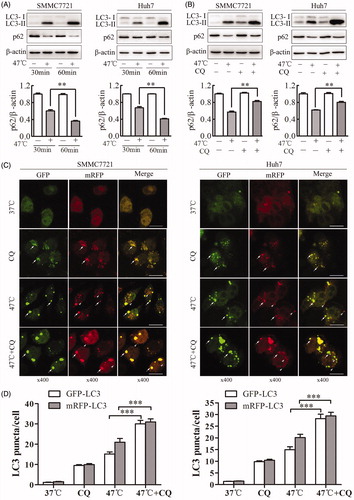
To clarify whether the accumulation of autophagosomes induced by sublethal heat stress is a result of the induction of autophagosome formation or the inhibition of autophagosome degradation, we measured the amount of SQSTM1/p62, a cargo protein recognized as a marker of autophagic flux by western blot [Citation22]. Activation of autophagic flux lead to a decrease in p62 levels and an increase in LC3-II levels. When SMMC7721 or Huh7 cells were exposed to heat stress at 47 °C from 30 to 60 min, the amount of p62 decreased, while LC3-II levels increased (). When cells were exposed to heat stress in the presence or absence of the autophagy inhibitor CQ, LC3-II levels were further increased in the combination group than in the heat-treated alone group, while p62 levels increased (). To confirm this finding, we examined the color changes of mRFP-GFP-LC3 puncta. SMMC7721 and Huh7 cells were transfected with mRFP-GFP-LC3 adenovirus and then exposed to sublethal heat stress. If an autophagosome fuses with a lysosome to form an autolysosome, GFP fluorescence but not mRFP fluorescence is attenuated under the acidic lysosomal conditions. As shown in , when cells were exposed to heat stress and CQ, the number of GFP puncta significantly increased, while the number of mRFP+/GFP − puncta decreased. This observation supports the notion that autophagy induced by sublethal heat stress proceeds to the lysosomal degradation phase. Our findings indicate that the accumulation of LC3-II induced by sublethal heat stress results from the induction of autophagosome formation, not from the blockade of autophagosome degradation.
Figure 2. Sublethal heat stress induces autophagic flux in HCC cells. (A, B) SMMC7721 and Huh7 cells were exposed to 47 °C for 30 or 60 min (A) or pretreated with the autophagy inhibitor CQ (20 μM) for 4 h and then exposed to 47 °C for 30 min (B). The expression levels of LC3-II and p62 were analyzed by western blot. p62 levels were quantified and normalized to those of β-actin. (C, D) Representative images and quantification results for LC3 puncta in SMMC7721 and Huh7 cells transiently transfected with mRFP-GFP-LC3 adenovirus followed by exposure to CQ (20 μM) for 4 h and heat treatment at 47 °C for 30 min. Arrows indicate co-localized GFP and mRFP puncta, while arrowheads indicate mRFP puncta. Scale bar, 20 μm. The average numbers of GFP-LC3 and mRFP-LC3 puncta per cell were counted in 6 random fields with at least 100 cells in each group. Each experiment was repeated three times, and representative images are shown. Error bars represent the SEM. **p < .01; ***p < .001.
Inhibition of autophagy enhances heat-induced apoptosis in HCC cells
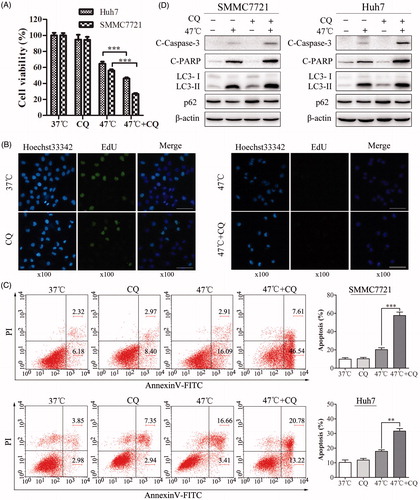
Accumulating evidence suggested a paradoxical role of autophagy in controlling cell survival and cell death under various conditions [Citation10,Citation18,Citation23,Citation24]. To determine the biological role of autophagy in heat-mediated apoptosis, the autophagy inhibitor CQ was used to disrupt lysosomal function and then block autophagic degradation. To this end, HCC cells were treated with CQ, heat stress or both in combination. Our results indicated that CQ enhanced the heat-induced suppression of cellular proliferation, as indicated by decreases in cell viability and EdU incorporation ( and Supplemental Figure S2(A)). In agreement with this observation, heat-induced apoptosis was markedly augmented in the presence of CQ, as indicated by an increased number of Annexin V-positive cells, as well as by the accumulation of cleaved Caspase-3 and PARP ().
Figure 3. Inhibition of autophagy by CQ markedly suppresses cell proliferation and enhances heat-induced apoptosis in HCC cells. (A) Cell viability was assessed by CCK-8 assays. SMMC7721 and Huh7 cells were pretreated with CQ (20 μM) for 4 h and then exposed to 47 °C for 30 min, followed by culture at 37 °C for 24 h. (B) SMMC7721 cells were pretreated with CQ (20 μM) for 4 h and then exposed to 47 °C for 30 min, followed by culture with EdU at 37 °C for 2 h. Images were taken under a fluorescence microscope. Scale bar, 20 μm. (C, D) SMMC7721 and Huh7 cells were treated as in (A), and apoptosis was analyzed by flow cytometry (C) and western blot (D). Each experiment was repeated three times and representative images are shown. Error bars represent the SEM. **p < .01; ***p < .001.
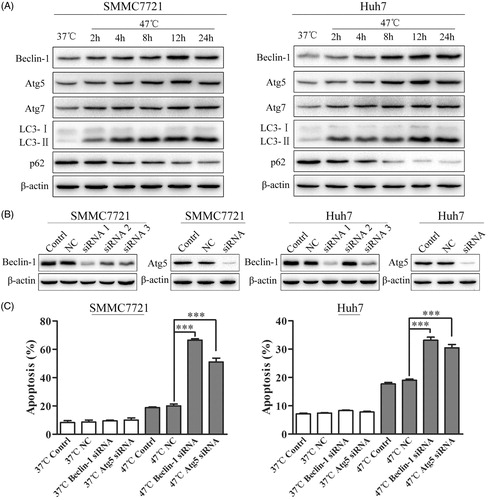
Considering that the pharmacological autophagy inhibitor CQ might have effects on lysosomes that are independent of autophagy, an alternative approach was applied to block the formation of autophagosomes via the knockdown of key autophagy genes, such as Beclin-1 and Atg5, using siRNA [Citation18]. First, we confirmed that heat treatment increased Beclin-1, Atg5, Atg7 and LC3-II levels and decreased p62 levels (). Furthermore, we found that silencing Beclin-1 and Atg5 significantly reduced Beclin-1 and Atg5 levels and markedly enhanced the heat-induced apoptosis of HCC cells (). Collectively, these results indicate that heat-induced autophagy plays a protective role against heat-induced apoptosis in HCC cells.
Figure 4. Inhibition of autophagy by Beclin-1 or Atg5 siRNA enhances heat-induced apoptosis in HCC cells. (A) SMMC7721 and Huh7 cells were exposed to 47 °C for 30 min followed by culture at 37 °C for the indicated time. The expression levels of Beclin-1, Atg5, Atg7, LC3-II and p62 were analyzed by western blot. (B) The efficacy of siRNA targeting Beclin-1 and Atg5 was measured by western blot. SMMC7721 and Huh7 cells were transfected with NC siRNA, Beclin-1 siRNA, or Atg5 siRNA for 48 h and then exposed to heat treatment, followed by recovery for 24 h. (C) SMMC7721 and Huh7 cells treated as in (B) were stained with 5 μL of FITC-Annexin V and 1 μL of 100 μg/mL PI working solution for 15 min in the dark. Apoptotic cells were analyzed by flow cytometry within 1 h. Each experiment was repeated three times and representative images are shown. Error bars represent the SEM. ***p < .001.
The ATP-AMPK-mTOR signaling pathway is involved in heat-induced autophagy in HCC cells
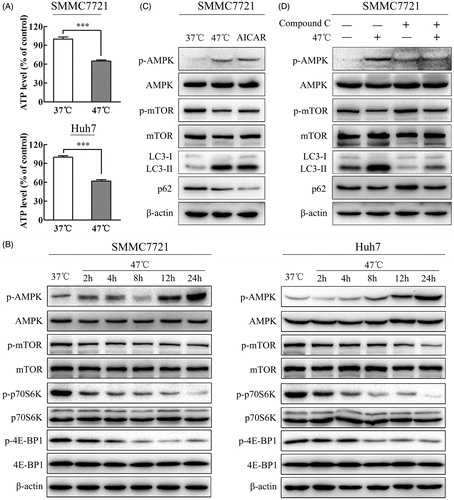
Previous studies showed that heat shock of rat hepatocytes could decrease ATP content, increase the AMP/ATP ratio, and lead to the activation of AMPK by phosphorylation [Citation25]. AMPK is an αβγ heterotrimer activated by decreasing concentrations of ATP and increasing AMP concentrations. In agreement with this finding, sublethal heat stress reduced the ATP level. As the level of ATP decreased, increasing concentrations of ADP resulted in an increase in AMP due to the adenylate kinase equilibrium [Citation26]. Thus, we speculated that the AMP/ATP ratio would increase and activate the AMPK signaling pathway. Our results revealed that sublethal heat stress increased AMPK phosphorylation and decreased the mTOR downstream targets p-p70S6K and p-4E-BP1 in a time-dependent manner, suggesting that the ATP-AMPK-mTOR signaling pathway plays an important role in heat-induced autophagy (). AMPK induces autophagy via the activation of ULK1 or the inactivation of mTOR [Citation27]. To further validate whether AMPK was involved in autophagy induced by heat stress, the effects of the AMPK activator AICAR and the AMPK inhibitor compound C on autophagy were analyzed in heat-treated cells. Similar to heat stress, AICAR activated AMPK via phosphorylation at Thr172 and induced an increase in LC3-II and a decrease in p62 ( and Supplemental Figure S3(A)), whereas compound C blocked the effects of heat stress-induced AMPK activation and autophagy ( and Supplemental Figure S3(B)). Taken together, these results indicate that the ATP-AMPK-mTOR signaling pathway is involved in heat-induced autophagy in HCC cells.
Figure 5. Heat stress induces autophagy through the ATP-AMPK-mTOR signaling pathway. (A) Heat stress decreased ATP in HCC cells after exposure to 47 °C for 30 min and recovery for 24 h. (B) Heat stress activated the AMPK-mTOR signaling pathway in SMMC7721 and Huh7 cells. The levels of AMPK, p-AMPK, mTOR, p-mTOR, p70S6K, p-p70S6K, 4E-BP1 and p-4E-BP1 were analyzed by western blot. SMMC7721 and Huh7 cells were exposed to 47 °C for 30 min, followed by recovery for 24 h. (C, D) The levels of p-AMPK, AMPK, p-mTOR, mTOR, LC3 and p62 were assessed by western blot. SMMC7721 cells were exposed to 47 °C for 30 min, followed by treatment with the AMPK agonist AICAR (1 mM) or the AMPK inhibitor compound C (10 μM) for 24 h. Each experiment was repeated three times, and representative images are shown. Error bars represent the SEM. ***p < .001.
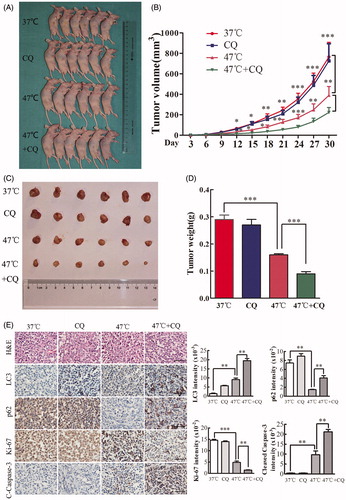
The autophagy inhibitor CQ enhances the antitumor effect of heat treatment in vivo
The autophagy inhibitor CQ disrupts lysosomal function and prevents the completion of autophagy [Citation21]. To examine whether CQ can potentiate the antitumor effect of heat treatment in vivo, we established a xenograft tumor model of SMMC7721 cells and tested the effects of vehicle, CQ, heat treatment alone, and a combination of CQ and heat treatment on tumor growth in nude mice. Nude mice were treated with vehicle or CQ three times a week. By day 30, there was no significant difference between these two groups (). Tumor growth was moderately inhibited by heat treatment (47 °C) but more significantly inhibited by the combination treatment (). Analysis of tumor weight at the end of the experiment indicated a statistically significant difference between heat treatment and the combination treatment (). IHC staining revealed that heat treatment significantly increased the expression of LC3 (a marker of autophagy) and decreased the expression of p62 (a marker of autophagic flux) (). These results indicate that heat treatment induces autophagy and autophagic flux in vivo. In addition, compared with heat treatment alone, heat treatment plus CQ markedly increased cleaved Caspase-3 levels but decreased Ki-67 expressions (). Taken together, these results indicate that targeting autophagy with CQ sensitizes cells to heat-induced apoptosis in vivo.
Figure 6. Inhibition of autophagy by CQ significantly enhances the antitumor effects of heat treatment in vivo. (A–D) SMMC7721 cells pretreated at 47 °C for 30 min or maintained at 37 °C were subcutaneously injected into the upper right flank of nude mice, treated with or without CQ and tumor size was measured. Mice were sacrificed 30 d after implantation (A). Tumor growth was measured every 3 d for 30 d, and the results were plotted (B). Tumors were harvested, photographed (C) and weighed, and the results were plotted (D). (E) Representative images of H&E and IHC staining. The levels of LC3, p62, Ki-67 and cleaved Caspase-3 were examined by IHC staining and quantified with Image-Pro Plus software. Scale bar, 100 μm. Each experiment was repeated three times and representative images are shown. Error bars represent the SEM. *p < .05, **p < .01 and ***p < .001.
Discussion
RFA has been accepted as a curative treatment alternative to surgical resection and liver transplantation for early-stage HCC [Citation6,Citation28]. However, the long-term prognosis of HCC patients treated with RFA is not satisfactory due to the high incidence of local recurrence [Citation29]. Thus, it is of great significance to explore the mechanisms underlying residual tumor progression and local recurrence of HCC after IRFA treatment.
To simulate the marginal zone of IRFA, HCC cells were exposed to sublethal heat stress at various temperatures ranging from 37 to 47 °C for different durations according to previous studies [Citation5,Citation6,Citation30]. Our results revealed that heat exposure induced autophagy in HCC cells in a temperature- and time-dependent manner. This phenomenon was particularly obvious at 47 °C for 30 min. Therefore, heat treatment at 47 °C for 30 min was selected for subsequent experiments. However, in this study, we found that cell proliferation was significantly suppressed after sublethal heat stress, which contradicts previous finding [Citation14]. This discrepancy may be due to differences in experimental models and procedures. As in previous studies, cells were cultured under hypoxic conditions to stimulate IRFA. However, in our study, cells were exposed to 47 °C for 30 min. Hypoxia cannot fully simulate the microenvironment of IRFA in clinic, while sublethal heat stress in a water bath may be a good experimental model to simulate both the hypoxic and hyperthermal conditions of IRFA. Further studies are needed to confirm this possibility.
Autophagy is an important mechanism to recycle energy and nutrients in response to starvation and environmental stresses [Citation9]. The role of autophagy in promoting cell survival or inducing cell death during the development of HCC is still controversial [Citation10]. Some previous studies showed that autophagy could promote HCC cell survival in response to environmental stresses [Citation18,Citation31], while other studies reported that autophagy induced HCC cell death [Citation32,Citation33]. Therefore, whether autophagy promotes cell survival or induces cell death depends on the cell type and environmental stress. Our team previously reported that IRFA promotes HCC cell progression via autophagy and the CD133 feedback loop [Citation13]. In this study, we found that inhibition of heat stress-induced autophagy enhances heat-induced apoptosis both in vitro and in vivo. In these two studies, we both reported that IRFA induces autophagy and autophagy flux in HCC cells, and IRFA promotes HCC cell survival via autophagy. In our previous study, we emphasized on revealing that IRFA increases the expression of CD133, a liver cancer stem cell marker, and IRFA-induced autophagy contributes to aggressive recurrence via the promotion of tumor progenitor characteristics. In this study, we reported a novel mechanism of heat induced-autophagy involved in the recurrence of HCC after IRFA. We found that heat-induced autophagy promotes HCC cell survival via the ATP-AMPK-mTOR signaling pathway, and inhibition of autophagy significantly enhances heat-induced apoptosis. Our new findings may be benefit for further understanding the role of autophagy in residual tumor progression and rapid local recurrence of HCC after IRFA treatment.
To the best of our knowledge, this study is the first to reveal that heat-induced autophagy plays a protective role against heat-induced apoptosis. The inhibition of autophagy by CQ or siRNA targeting Beclin-1 or Atg5 enhanced heat-induced apoptosis both in vitro and in vivo. In addition, we found that SMMC7721 cells were more sensitive than Huh7 cells to heat-induced apoptosis. This phenomenon can be explained by differences in p53 status between these two cell lines: SMMC7721 cells contain wild-type p53, while Huh7 cells express mutant p53 [Citation34,Citation35].
Autophagy can be regulated by various signaling pathways. Previous studies reported that heat-induced autophagy is activated by HSF1 [Citation36]. Heat shock protein 27, 70 and 90 levels are significantly increased after sublethal heat treatment [Citation5]. The heat shock response and autophagy coordinate to maintain proteostasis in eukaryotic systems [Citation37]. Zhao et al. demonstrated that HIF-1α is involved in IRFA-induced autophagy [Citation14]. In this study, we found that a novel pathway, the ATP-AMPK-mTOR signaling pathway, was involved in heat-induced autophagy. From the viewpoint of energy stress, our results demonstrated that sublethal heat stress reduced ATP levels. As ATP levels decreased, increasing concentrations of ADP resulted in an increase in AMP due to the adenylate kinase equilibrium [Citation26]. Thus, we speculated that sublethal heat stress reduced ATP levels, leading to an increase in the AMP/ATP ratio and, ultimately, activation of the AMPK-mTOR signaling pathway. These results were further confirmed by the effects of the AMPK activator AICAR and the AMPK inhibitor, compound C.
The role of autophagy in tumorigenesis and tumor suppression remains controversial [Citation10,Citation23]. Many studies have indicated that autophagy plays a dual role in the development of HCC. Thus, understanding the precise role of autophagy in HCC is important for facilitating the development of targeted treatments for HCC [Citation31,Citation32]. To examine whether CQ can potentiate the antitumor effect of heat treatment in vivo, a xenograft tumor model was established according to previous studies [Citation5,Citation38,Citation39]. The results revealed that heat treatment activated autophagy and autophagic flux in xenograft tumor tissues. In addition, the inhibition of autophagy by CQ significantly suppressed tumor growth and enhanced heat-induced apoptosis in vivo. Therefore, our results indicate that the inhibition of heat-induced autophagy may be a potential target for improving the efficacy of RFA treatment for HCC. These results are of great clinical and translational importance.
Conclusions
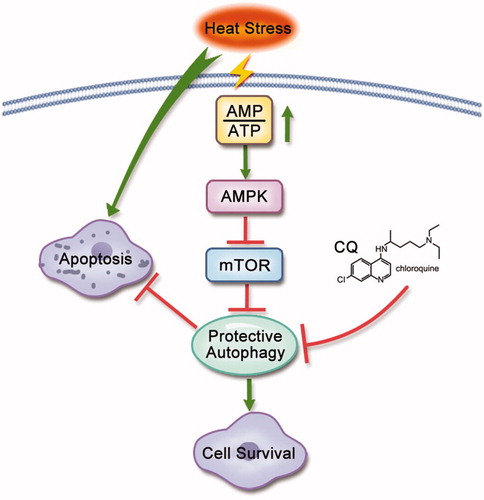
In conclusion, our results revealed that sublethal heat stress induced protective autophagy in HCC cells, both in vitro and in vivo. Furthermore, we demonstrated that the inhibition of autophagy by CQ or siRNA targeting Beclin-1 or Atg5 enhanced heat-induced apoptosis. Mechanistically, we demonstrated that the ATP-AMPK-mTOR signaling pathway was involved in heat-induced autophagy (). Our results suggest that targeting autophagy may be a promising therapeutic strategy to improve the efficacy of RFA treatment for HCC.
Figure 7. Schematic diagram of the mechanism of heat-induced autophagy that protects HCC cells from heat-induced apoptosis. Under heat treatment, ATP levels decrease, leading to an increase in the AMP/ATP ratio, which directly activates AMPKα by phosphorylation at Thr172 and then inactivates mTOR, leading to autophagy. Autophagy protects against heat treatment-triggered apoptosis. Inhibition of autophagy by CQ can enhance heat-triggered apoptosis. Arrows represent promotion events; blunt arrows indicate suppression events.
Supplemental Material
Download MS Word (29.5 KB)Supplemental Material
Download JPEG Image (413.1 KB){kind=link}
Supplemental Material
Download JPEG Image (146.6 KB){kind=link}
Supplemental Material
Download JPEG Image (486.6 KB){kind=link}
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;1;136:E359–E386.
- Wan MLY, El-Nezami H. Targeting gut microbiota in hepatocellular carcinoma: probiotics as a novel therapy. Hepatobiliary Surg Nutr. 2018;7:11–20.
- Nagarajan VK, Gogineni VR, White SB, et al. Real time evaluation of tissue optical properties during thermal ablation of ex vivo liver tissues. Int J Hyperthermia. 2018;35:176–182.
- Ke S, Ding XM, Kong J, et al. Low temperature of radiofrequency ablation at the target sites can facilitate rapid progression of residual hepatic VX2 carcinoma. J Transl Med. 2010;8:73.
- Yoshida S, Kornek M, Ikenaga N, et al. Sublethal heat treatment promotes epithelial-mesenchymal transition and enhances the malignant potential of hepatocellular carcinoma. Hepatology. 2013;58:1667–1680.
- Kong J, Kong J, Pan B, et al. Insufficient radiofrequency ablation promotes angiogenesis of residual hepatocellular carcinoma via HIF-1alpha/VEGFA. PLoS One. 2012;7:e37266.
- Kong J, Kong L, Kong J, et al. After insufficient radiofrequency ablation, tumor-associated endothelial cells exhibit enhanced angiogenesis and promote invasiveness of residual hepatocellular carcinoma. J Transl Med. 2012;10:230.
- Mizushima N, Levine B, Cuervo AM, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075.
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293.
- Lee YJ, Jang BK. The role of autophagy in hepatocellular carcinoma. Int J Mol Sci. 2015;16:26629–26643.
- Wang K, Liu R, Li J, et al. Quercetin induces protective autophagy in gastric cancer cells: involvement of Akt-mTOR- and hypoxia-induced factor 1α-mediated signaling. Autophagy. 2011;7:966–978.
- Zhao Y, Gong S, Shunmei E, et al. Induction of macroautophagy by heat. Mol Biol Rep. 2009;36:2323–2327.
- Wang X, Deng Q, Feng K, et al. Insufficient radiofrequency ablation promotes hepatocellular carcinoma cell progression via autophagy and the CD133 feedback loop. Oncol Rep. 2018;40:241–251.
- Zhao Z, Wu J, Liu X, et al. Insufficient radiofrequency ablation promotes proliferation of residual hepatocellular carcinoma via autophagy. Cancer Lett. 2018;1;421:73–81.
- Wang X, Liu J, Zhen J, et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 2014;86:712–725.
- Liu H, Ma Y, He HW, et al. SLC9A3R1 stimulates autophagy via BECN1 stabilization in breast cancer cells. Autophagy. 2015;11:2323–2334.
- Yu T, Guo F, Yu Y, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell. 2017;170:548–563 e16.
- Ding ZB, Hui B, Shi YH, et al. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clin Cancer Res. 2011;17:6229–6238.
- Kou JY, Li Y, Zhong ZY, et al. Berberine-sonodynamic therapy induces autophagy and lipid unloading in macrophage. Cell Death Dis. 2017;8:e2558.
- Xie CM, Liu XY, Sham KW, et al. Silencing of EEF2K (eukaryotic elongation factor-2 kinase) reveals AMPK-ULK1-dependent autophagy in colon cancer cells. Autophagy. 2014;10:1495–1508.
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326.
- Liu H, Ma Y, He HW, et al. SPHK1 (sphingosine kinase 1) induces epithelial-mesenchymal transition by promoting the autophagy-linked lysosomal degradation of CDH1/E-cadherin in hepatoma cells. Autophagy. 2017;13:900–913.
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995.
- Pei G, Luo M, Ni X, et al. Autophagy facilitates metadherin-induced chemotherapy resistance through the AMPK/ATG5 pathway in gastric cancer. Cell Physiol Biochem. 2018;46:847–859.
- Corton JM, Gillespie JG, Hardie DG. Role of the AMP-activated protein kinase in the cellular stress response. Curr Biol. 1994;4:315–324.
- Oakhill JS, Steel R, Chen ZP, et al. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011;332:1433–1435.
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945.
- Tan Y, Jiang J, Wang Q, et al. Radiofrequency ablation using a multiple-electrode switching system for hepatocellular carcinoma within the Milan criteria: long-term results. Int J Hyperthermia. 2018;34:298–305.
- Feng X, Xu R, Du X, et al. Combination therapy with sorafenib and radiofrequency ablation for BCLC Stage 0-B1 hepatocellular carcinoma: a multicenter retrospective cohort study. Am J Gastroenterol. 2014;109:1891–1899.
- Li Y, Brown RE, Martin RC. Incomplete thermal ablation of hepatocellular carcinoma: effects on tumor proliferation. J Surg Res. 2013;181:250–255.
- Xiong H, Ni Z, He J, et al. LncRNA HULC triggers autophagy via stabilizing Sirt1 and attenuates the chemosensitivity of HCC cells. Oncogene. 2017;36:3528–3540.
- Han B, Yu YQ, Yang QL, et al. Kaempferol induces autophagic cell death of hepatocellular carcinoma cells via activating AMPK signaling. Oncotarget. 2017; 8:86227–86239.
- Yao C, Liu BB, Qian XD, et al. Crocin induces autophagic apoptosis in hepatocellular carcinoma by inhibiting Akt/mTOR activity. OncoTarget Ther. 2018;11:2017–2028.
- Shimizu S, Takehara T, Hikita H, et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J Cancer. 2012;131:548–557.
- Gao Y, Lin LP. Growth arrest induced by C75, A fatty acid synthase inhibitor, was partially modulated by p38 MAPK but not by p53 in human hepatocellular carcinoma. Cancer Biol Ther. 2006;5:978–985.
- Kumsta C, Chang JT, Schmalz J, et al. Hormetic heat stress and HSF-1 induce autophagy to improve survival and proteostasis in C. elegans. Nat Comms. 2017;8:14337.
- Zhang Y, Calderwood SK. Autophagy, protein aggregation and hyperthermia: a mini-review. Int J Hyperthermia. 2011;27:409–414.
- Dong S, Kong J, Kong F, et al. Insufficient radiofrequency ablation promotes epithelial-mesenchymal transition of hepatocellular carcinoma cells through Akt and ERK signaling pathways. J Transl Med. 2013;11:273.
- Dong S, Kong J, Kong F, et al. Sorafenib suppresses the epithelial-mesenchymal transition of hepatocellular carcinoma cells after insufficient radiofrequency ablation. BMC Cancer. 2015;15:939.