
Abstract
The success of melanoma immunotherapy is dependent on the presence of activated and functional T-cells in tumors. The objective of this study was to investigate the impact of local-focused ultrasound (FUS) heating (∼42–45 °C) and in-situ anti-CD-40 agonistic antibody in enhancing T-cell function for melanoma immunotherapy. We compared the following groups of mice with bilateral flank B16 F10 melanoma: (1) Control, (2) FUS, (3) CD-40, and (4) CD-40 + FUS (FUS40). FUS heating was applied for ∼15 min in right flank tumor, and intratumoral injections of CD-40 were performed sequentially within 4 h. A total of 3 FUS and 4 anti-CD-40 treatments were administered unilaterally 3 days apart. Mice were sacrificed 30 days post-inoculation, and the treated tumor and spleen tissues were profiled for T-cell function and macrophage polarization. Compared to all other groups, histology and flow cytometry showed that FUS40 increased the population of tumor-specific CD-4+ and CD-8+ T cells rich in Granzyme B+, interleukin-2 (IL-2) and IFN-γ production and poor in PD-1 expression. In addition, FUS40 promoted the infiltration of tumor-suppressing M1 phenotype macrophages in the treated mice. The resultant immune-enhancing effects of FUS40 suppressed B16 melanoma growth at the treated site by 2–3-folds compared to control, FUS, and CD-40, and also achieved significant abscopal effects in untreated tumors relative to CD40 alone. Additionally, the local FUS40 prevented adverse liver toxicities in the treated mice. Our study suggests that combined FUS and CD-40 can enhance T-cell and macrophage functions to aid effective melanoma immunotherapy.
Introduction
Malignant melanoma is a highly metastatic and often lethal cancer, and incidence rates continue to rise steadily [Citation1]. Most melanoma patients with metastatic disease are resistant to chemo- and radiotherapy and median survival rates are typically <4years. Immunotherapy using antibodies that block CTLA-4, PD-1, and PDL1 to activate anti-tumor immunity has improved outcomes in a subset of patients [Citation2,Citation3]. This is a highly promising strategy, and depending on the tumor microenvironment, expression of target proteins, and cancer types can generate a response rate of 10-50% [Citation4]. Despite profound clinical benefits for some, a large proportion (>50%) of melanoma patients do not respond to the immunotherapy. This is attributed to a lack of a baseline T-cell infiltration, and presence of dysfunctional T-cells characterized by an enhancement of PD-1 inhibitory functions and reduced Interleukin-2 (IL-2), Granzyme B and IFN-γ cytokine production [Citation5]. Thus, new approaches are needed to prevent immune cell dysfunctions and T-cell exhaustion for effective immunotherapy. Towards this goal, this study investigated the role of locally applied focused ultrasound (FUS) heating (∼42–45 °C) and in-situ (intratumoral) injection of anti-CD-40 agonistic antibody in augmenting T-cell and macrophage functions for local and systemic immunity against murine melanoma. In-situ vaccination compared to systemic therapies utilize all relevant antigens, whether tumor-associated or neoantigens to generate robust antitumor response, thereby eliminating the need to identify and isolate the tumor antigens for adaptive immunity [Citation6,Citation7].
CD-40 is a member of the tumor necrosis factor receptor family and is highly expressed in antigen presenting cells (APCs) including macrophages, monocytes, dendritic cells, and B cells [Citation8,Citation9]. Under normal conditions, T-helper cells expressing CD-40 ligand (CD-40L, CD154) can interact with APCs via CD-40, resulting in enhanced antigen-presentation and release of proinflammatory cytokines [Citation10–14]. Some studies have also shown that the systemic administration of CD-40 agonists lowers the intratumoral PD-1 expression in T-cells, and aid the phenotypic conversion of macrophages from M2 to M1 [Citation15–17]. Currently, several clinical trials are investigating the role of anti-CD40 in various tumor types (NCT02376699, NCT03389802, NCT03123783, NCT03597282 NCT03165994, NCT02706353) [Citation18,Citation19]. FUS-induced local heating and associated stress can modify the tumor cells and microenvironment, causing antigen release, expression of heat-shock proteins, upregulation of pro-phagocytic signals such as calreticulin (CRT), and overall stimulate tumor immunity. Unlike ionizing radiation, which damages collateral tissues and induces oncogenic proteins, FUS generates protein coagulation and non-lethal thermal stress in less aggressively treated tumors [Citation20,Citation21]. Although radiation combined CD40 studies are starting to emerge [Citation22,Citation23], not much is currently known about how anti-CD40 synergises with FUS heating. Here, murine melanoma treated locally with CD-40 and FUS were profiled for the polarization status of macrophages and T-cell phenotypes. Data suggest that the combined CD-40 and FUS can prevent T-cell dysfunction and exhaustion, and improve macrophage polarization dynamics, suggesting the value of the proposed combinatorial modality in melanoma immunotherapy.
Materials
B16F10 murine melanoma cells were provided by Dr. Mary Jo Turk at Geisel School of Medicine at Dartmouth (Hanover, NH). B16F10 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% streptomycin/penicillin. Anti-CD-40 agonist antibody (FGK45) was purchased from BioXCell (West Lebanon, NH, USA). Fluorochrome-conjugated monoclonal antibodies (mAbs) for flow cytometry were purchased from BioLegend (San Diego, CA) and are listed here: APC anti-CD-4 (GK1.5), PE anti-CD3 (145-2C11), BB515 anti-MHCII (2G9), APC-Cy7 anti-IFN-γ (XMG1.2), APC-Cy7 anti-CD11c (1A8), FITC anti-CD-45.2 (104), PE anti-Granzyme B (QA16A02), APC anti-CD206 (C068C2), PE anti-CD11b (M1/70), and Pe-Cy7 anti-IL-2 (JES6-5H4) and anti-CD16/CD32 (Clone 93). Alexa Fluor 700 or Pe-Cy7 anti-CD-45 (30-F11), BV480 anti-F4/80 (T45-2342), V500 anti-CD3 (500A2), BV786 anti-CD-4, APC-H7 anti-CD-8a (53-6.7), BV650 anti-IFN-γ (XMG1.2), and Alexa Fluor 488 anti-Foxp3 (MF23) were purchased from BD Biosciences (San Jose, CA).
Mouse melanoma model generation and study design
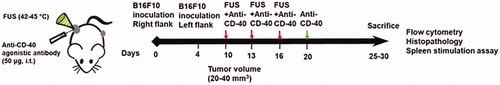
All animal-related procedures were approved and carried out under the guidelines of the Oklahoma State University Animal Care and Use Committee. We compared the following groups (n = 6): 1) Control, 2) FUS, 3) CD-40, and 4) FUS + CD-40 (FUS40). 0.5 × 106 B16F10 cells in 50 μL of PBS was injected subcutaneously (sc) in the right flank regions of C57/BL6 mice. 4 days later, the mice were injected with 0.125 × 106 cells in the left flank region by sc route. Mice tumor volumes were measured daily by serial caliper measurements using the formula (length × (width)2)/2, where length was the largest dimension and width was the smallest dimension perpendicular to the length. Unilateral treatment of the right flank tumor was initiated at a volume of 20-40 mm3. FUS heating (42-450C) was applied for ∼15min, and intratumoral injections of CD-40 antibody (50 μg/session) was performed sequentially within 4 h after FUS heating (). A total of 3 FUS and 3 anti-CD-40 treatments 3 days apart was performed. Additionally, on day 20 post inoculation, CD-40 alone was administered in the mice. Mice were sacrificed when the tumors reached >1cm in any dimension or 30 days post-inoculation. The right flank tumors and the spleen from the euthanized mice were excised, weighed, and processed for flow cytometry and histopathological studies. For flow cytometry, two-thirds of the harvested tumor was processed immediately. Specifically for flow studies, tumor samples (n = 5/group) and spleen (n = 4-5/group) were randomly selected and processed for immune cell profiling. For histopathological analysis, the remaining one-third of the tumor tissue was fixed in 10% neutral buffered formalin. Blood samples (n = 6) were also collected by intracardiac route for biochemical analysis of liver function.
Figure 1. Experimental design to assess the efficacy of FUS and CD-40 combination against melanoma tumors. 0.5 × 106 B16F10 cells were injected subcutaneously (sc) in the right flank regions of C57/BL6 mice. 4 days later, the mice were injected with 0.125 × 106 cells in the left flank region by sc route. Unilateral treatment of the right flank tumor was initiated at a volume of 20–40 mm3. FUS heating (42–45°C) was applied for ∼15 min, and intratumoral injection of anti-CD-40 agonistic antibody (50 µg) was performed sequentially within 4 h of FUS heating. Red arrows indicate the three treatments with FUS and CD-40. Green arrow indicates the fourth anti-CD-40 dose. Mice were sacrificed when tumors reached >1 cm in any dimension or reached 30 days post-inoculation. The harvested treated tumor and spleen were analyzed for the population and type of immune cell.
FUS set-up and treatment methodology
All FUS tumor treatment was performed using an Alpinion transducer with a 1.5 MHz central frequency, 45 mm radius, and 64 mm aperture diameter with a central opening of 40 mm in diameter. For FUS exposure, the center of the tumor was aligned at a fixed focal depth for efficient coverage (voxel size: 5 × 5 × 12 mm), and the alpinion VIFU-2000 software was used to define target boundary and slice distance in x, y, and z directions for automatic rastering of the transducer for 15 min. As the tumor grew, the focal point was rastered to cover the entire tumor. FUS treatment parameters used were as follows: 5 Hz frequency, 50% duty cycle, and 6 W acoustic power. The combination of these parameters achieved a mean target temperature of 42–45 °C at the focus (measured by inserting a fiber optic temperature sensor; Qualitrol, Quebec, Canada) inside the tumor ().
Immunophenotyping of melanoma tumors with flow cytometry
Single-cell suspensions obtained from the mechanical disruption of the tumors (n = 5 mice/group) followed by enzymatic digestion with 200 U/mL collagenase IV (Life Technologies, NY, USA) were filtered through a 70-μm cell strainer (Corning Inc, Corning, NY). Cell suspensions were stained using the fixable viability stain 575 V (BD Biosciences) according to the manufacturer’s instructions to exclude dead cells and anti-CD16/CD32 antibody to block FcγIII/II receptor-mediated unspecific binding. The following panel of the indicated fluorochrome-conjugated anti-mouse antibodies were used to stain cells for 30 min in dark on ice: CD-45+ (Tumor infiltrating leukocytes; TILs), CD3+, CD-4+ (CD-4+ T or helper Th cells), CD3+, CD-8+ (CD-8+ T cells), CD11b+, F4/80+ (macrophages), CD11b+, F4/80+, MHCII high (MHCII high M1 macrophages), and CD11b+, F4/80+ MHCII lo/neg, CD206+ (M2 macrophages). For detecting IL-2, IFN-γ, Granzyme-B, and Foxp3 positive Treg cells, cells were washed after surface marker staining, fixed and permeabilized with transcription factor buffer set (BD Biosciences, San Jose, CA) and incubated with Pe-Cy7 anti-IL-2, BV650 or APC-Cy7 anti-IFN-γ, PE anti-Granzyme-B or Alexa Fluor 488 anti-Foxp3 antibody for 30 min in the dark on ice. Stained cells were run in LSRII analyzer (BD Biosciences) within 24 h. Compensations were performed with single-stained UltraComp eBeads or cells (). Datasets were analyzed using FlowJo software v.10.2 (Treestar Inc, Ashland, OR, USA). For all channels, positive and negative cells were gated on the basis of fluorescence minus one control.
Characterization of the T-cell activity and melanoma-specific systemic immunity
Single cell suspension of splenocytes (n = 4–5) were stimulated ex-vivo with melanoma-specific differentiation antigen tyrosinase-related protein 2 (TRP-2) peptide for 10–12 h to evaluate generation of TRP-2 melanoma antigen-specific immunity [Citation24,Citation25]. Briefly, 1–2 × 106 splenocytes were incubated with 2.5 µg TRP-2 peptide for 10–12 h in the presence of Brefeldin A (eBioscience, 1000X solution) at 37 °C and 5% CO2. Treated cells were washed with PBS and stained with CD-45, CD3, CD-4, CD-8, IL-2 and IFN-γ antibodies for flow cytometry. The number of T-effector (Teff) responding to TRP-2 stimulation was calculated as CD-45+ CD3+ CD-4+ or CD-8+ T cells that were positive for IFN-γ or IL-2. Data were expressed as the percentage of the total splenocytes.
Histopathological analysis of treated tumors
The control, FUS, CD-40, and FUS/CD-40 tumor tissues (n = 5) were fixed in 10% neutral buffered formalin, processed, and embedded in paraffin as previously described [Citation26]. Histopathological examination was made on sections (4 μm) stained with hematoxylin and eosin (HE). The tumor sections were screened qualitatively for immune infiltration using an Olympus BX50 microscope with Olympus DP26 digital photography by a veterinary pathologist blinded to treatment groups. These findings were also validated by quantitative flow cytometry assessment of tumor infiltrating leukocytes in the tumor samples (n = 5).
Hepatotoxicity assessment of serum samples from the treated mice
Serum samples (n = 6/group) from mice that reached study endpoints were analyzed by Dr. Charles Wiedmeyer from Comparative Clinical Pathology Services (Columbia, MO) for the liver function test. Specifically, Alanine aminotransferase (ALT), Aspartate aminotransferase (AST) and albumin to globulin ratio were evaluated to assess liver function.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 8.0 software (GraphPad Software Inc, La Jolla, CA, USA). Data are presented as mean ± SEM unless otherwise indicated. For analysis of 3 or more groups, a one-way ANOVA test was performed followed by Fisher’s LSD without multiple comparisons correction. Analysis of differences between 2 normally distributed test groups was performed using an unpaired t-test assuming unequal variance. p values less than .05 were considered significant.
Results
FUS40 enhanced survival and delayed tumor growth rates in treated and untreated sites
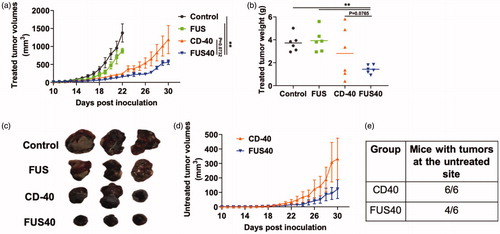
The treated flank tumor volumes in mice were monitored over 30 days post-inoculation (pi). Both control and FUS treated tumors showed a progressive increase in the tumor volumes in the treated site and reached sacrifice end-points (>1cm in any dimension or >15% loss in the body weight) by day 21 pi. In contrast, CD-40 and FUS40 achieved significant growth delay at the treated site. That said, FUS40 most effective amongst all the treatment groups (∼2–3-fold > tumor regression compared to control, FUS, and CD-40; . FUS40 also decreased tumor weight to a significantly greater extent by visual and statistical measures compared to all other groups (. We next compared abscopal effects in the contralateral untreated site. As control and FUS mice reached sacrifice endpoint early in the trial, they were not included for the enumeration of systemic immune-effects. Data showed that FUS40 induced superior regression of untreated tumor volumes over 30 days compared to CD40 alone (. Furthermore, two out of six FUS40 treated mice demonstrated systemic immunity against tumor challenge. In contrast, CD-40 treated mice demonstrated a 100% tumor take at the untreated side (.
Figure 2. Local FUS therapy and in situ anti-CD-40 agonistic antibody suppressed the tumor growth of local and distant untreated site in B16F10 melanoma model. (a) Mean volumes of the treated tumors are shown till 30 days. Control and FUS reached sacrifice end points by day 21. CD-40 and FUS40 significantly decreased tumor volumes compared to FUS and untreated tumors; (b) Tumor weights at the time of sacrifice showed a significant reduction in the overall weight for FUS40 compared to other groups. (c) Representative images of the treated tumor. (d) Mean volumes of the distant untreated tumors are shown till 30 days. (e) Number of mice that were tumor free at the distant untreated site. Results are shown as mean ± SEM. One-way ANOVA followed by Fisher’s LSD without multiple comparisons correction. *p < .05, **p < .01.
FUS40 promoted the recruitment of tumor infiltrating leukocytes (TILs) and granzyme B + PD-1- CD-8 +T- cells in treated tumors
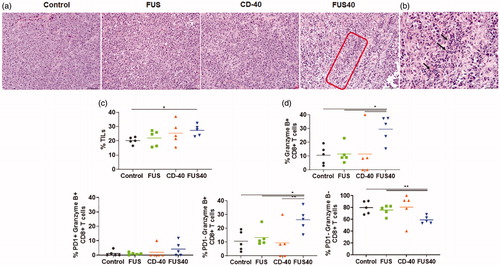
Analysis of tumor sections by H&E staining revealed prominent multifocal regions of coagulative necrosis in treated tumors compared to untreated control (. FUS40-treated tumors exhibited significantly higher levels of perivascular infiltration of lymphocytes within the tumor mass and the presence of CD-45 expressing leukocyte in histology and flow cytometry among all the groups (). To further characterize the functional status of the infiltrated immune cells, the CD-8+ T-cells were probed for Granzyme B + and PD-1+ expression by flow cytometry. We found that FUS40 promoted an activated Granzyme B + PD-1 CD-8+ T-cells phenotype and these were 2-fold higher than other groups (. In contrast, the control, FUS, and CD-40 tumors were primarily composed of PD-1+ Granzyme B- or non-activated PD-1- Granzyme B- phenotypes, indicating that the functional status of CD-8+ T-cells was likely preserved by FUS40 therapy.
Figure 3. FUS40 enhanced the recruitment of leukocytes and prevented T-cell dysfunction. (a) Compared to other groups, FUS40-treated tumors exhibited relatively higher perivascular infiltration of lymphocytes (red box) within the tumor mass upon qualitative imaging by a veterinary pathologist blinded for the groups; n = 5, Hematoxylin:Eosin stain, Bar = 50 μm. (b) Enlarged view of FUS40 tumor sections (red box) showing perivascular infiltration of lymphocytes (black arrows), Bar = 20 μm. Differences were analyzed by an unpaired t test assuming unequal variance. (c) Flow cytometry showed that the frequency of tumor infiltrating leukocytes in FUS40 tumors was significantly greater than the control tumors. (d) Percentage of Granzyme-B + CD3+ CD8+ T cells was significantly higher for FUS40 (2–3-fold) compared to all other groups. FUS40 preserved activated CD8+ T cell from functional exhaustion by inhibiting PD-1 expression and enhancing Granzyme B production. For all channels, positive and negative cells were gated on the basis of fluorescence minus one control. Results are shown as mean ± SEM. *p < .05, Data were analyzed using a one-way ANOVA followed by Fisher’s LSD without multiple comparisons correction.
FUS40 enhanced the melanoma-specific production of IL-2 and IFN-γ from T-cells in the spleen
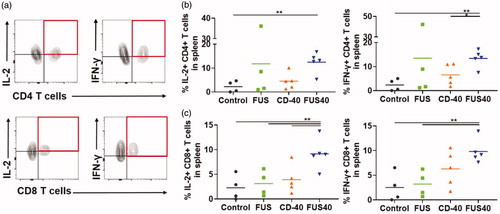
Dysfunctional and exhausted T-cells are not efficient in producing cytokines such as IL-2, TNF-α, and IFN-γ, or Granzyme B. Thus, to gain an understanding of the functional status of T-cells, the splenocytes were stimulated with the melanoma-specific TRP-2 peptide and assessed for the production of IL-2 and IFN-γ. A 2-fold higher expression of the cytokines was noted in the CD-4+ and CD-8 + T cells for FUS40 compared to CD-40, FUS, and control treatments ().
Figure 4. FUS40 revived the production of effector cytokines from melanoma specific CD4+ and CD8+ T cells in spleen. B16F10 melanoma bearing mice treated sequentially with FUS and anti-CD-40 agonistic antibody were sacrificed and spleen was evaluated for TRP-2 specific immunity in an ex vivo stimulation assay. (a) Flow cytometry contour plots representing the gating strategy for CD4+ and CD8+ T cells producing IL-2 and IFN-γ. (b) IL-2 and IFN-γ secreting CD4+ T cells in splenocytes after ex vivo TRP-2 stimulation were significantly increased by the FUS40 compared to control. Differences were analyzed by an unpaired t test assuming unequal variance. (c) The highest frequency of CD8+ T cells producing IL-2 and IFN-γ was observed in FUS40. *p < .05, **p < .01, one-way ANOVA followed by Fisher’s LSD without multiple comparisons correction.
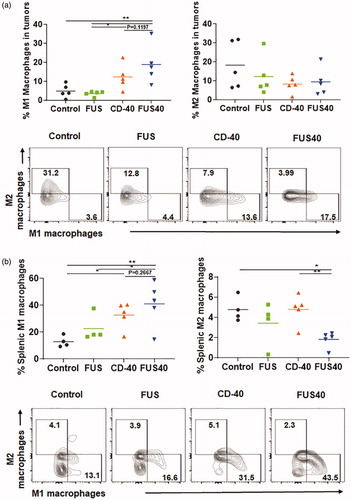
FUS and CD-40 promoted the M1 macrophage phenotype in the tumors and spleen without significantly altering T-reg populations
Tumor-associated macrophages (TAMs) are known to release cytokines and chemokines that generally suppress cytotoxic effects of CD-8+ T cell [Citation27,Citation28]. These suppressive cells are often referred to as M2 macrophages. One potential mechanism of immunotherapy is reducing the prevalence of immunosuppressive M2 macrophages and increasing immunostimulatory macrophages MHCII high expressing M1 phenotype cells can activate and restore T cell effector activity [Citation29,Citation30]. We analyzed the tumors and spleen for M1 and M2 macrophage populations. FUS40 resulted in a ∼1.3-2- fold enhancement of M1 phenotype compared to other groups in spleen and tumors (). The increase in M1 phenotype did not accompany an increase of M2 macrophages in the tumor. Additionally, the M2 macrophage was significantly decreased (∼2-2.5 fold) in the spleen with FUS40 compared to FUS and CD-40 alone (. Furthermore, the population of Tregs that infiltrate tumors in response to chemokines secreted by TAMs was found to be unchanged between various treatments (Figure S2) [Citation31].
Figure 5. FUS40 promoted M1 macrophage polarization in the tumor and the spleen. (a) Frequency of M1 macrophages in the tumor was increased by 4-fold for FUS40 compared to FUS and control, whereas M2 macrophages in treated tumors remained unaltered compared to controls. CD11b + F4/80+ MHCII high (M1 macrophages) and CD11b + F4/80+ MHCII lo/neg CD206+ (M2 macrophages). (b) An increased percentage of M1 macrophages was observed in the spleens from CD-40 and FUS40 cohorts. FUS40 reduced the frequency of M2 macrophages in the spleen compared to other groups. Data are shown as mean ± SEM. Statistics were determined by ANOVA followed by Fisher’s LSD without multiple comparisons correction. *p < .05, **p < .01.
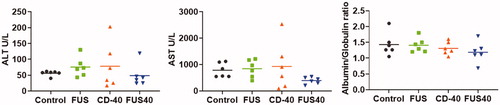
In-situ FUS40 treatment did not impair liver functions
Systemic anti-CD-40 agonist administration is known to cause immune-mediated hepatotoxicity [Citation32]. To assess whether intratumoral CD-40 impacted the liver functions, the ALT, AST, and albumin/globulin ratio in the treated mice sera were assessed. Both monotherapies (CD-40 or FUS) and combined FUS40 did not significantly alter the serum levels of liver enzymes and protein compared to untreated mice (.
Figure 6. Local FUS40 and CD-40 therapy did not cause liver toxicity in B16F10 melanoma bearing mice. Levels of ALT, AST, and Albumin to Globulin ratio in the serum of mice were determined at the time of sacrifice 25–30 days post tumor inoculation. Data were analyzed by ANOVA followed by Fisher’s LSD without multiple comparisons correction (n = 6).
Discussion
The success of melanoma immunotherapy is highly dependent on the type of tumor microenvironment [Citation33]. The objective of this study was to test whether combined FUS40 can modify key immune-suppressive pathways and stimulate immune effector pathways in melanoma tumors to promote local and systemic immunity. FUS-induced local heating and stress are known to modify the tumor microenvironment to enhance vascular permeability and infiltration of immune cells [Citation20,Citation34–43]. We hypothesized that FUS combined with enhanced immune infiltration combined with intratumoral agonistic anti-CD-40 antibody would enrich the populations of functional T-cells and macrophages, allowing superior protection against metastatic disease.
For evaluation of therapeutic and systemic immune effects, mice with bilateral tumors were exposed to FUS, CD-40 and combined FUS and CD-40 (FUS40) on the right flank tumor (. Monotherapy with FUS failed to improve survival rates compared to control. In contrast, CD-40 and FUS40 prolonged survival and suppressed the tumor growth rates at the treated sites (). Also, amongst all the groups, FUS40 was most potent at inducing tumor growth delay and abscopal effect at the untreated site, highlighting that the nonablative FUS dose can synergize with in-situ immune therapies ( and Figure S1). To determine if the induction of abscopal effects was mediated by the infiltration of cytotoxic T-cells, the treated tumor and spleen tissues were characterized for the production of IL-2, IFN-γ, and Granzyme B and the surface expressions of PD-1 [Citation28,Citation44]. Production of cytokines such as IL-2 from CD-4+ and CD-8+ T cells regulate the differentiation of T cells to Th1 cells, induce perforin, granzyme B, and IFN-γ production, and prevent T-cell exhaustion [Citation45,Citation46]. Results indicated that the splenocytes from the FUS and CD-40 treated mice that were stimulated with melanoma-specific TRP-2 antigen did not alter the IL-2 and IFN-γ productions from the CD4+ and CD8 + T cells. In contrast, the FUS40 treated mice achieved a 2-3 fold higher production of the cytokines as well as the expansion of the T-cells. To gain further understanding of the activation mechanisms, we next characterized the surface expression of PD-1 checkpoint protein and production of Granzyme B from the T-cells present in the treated tumor. Granzyme B is the key component of T-cell mediated tumor cell lysis [Citation47]. However, a higher expression of PD-1 expression can reduce Granzyme B effect and drive T-cells to an exhausted stage [Citation48]. We found that FUS40 consistently increased the proportion of Granzyme B + PD-1- CD-8+ T-cells in the treated tumors () compared to the control, FUS and CD-40 treated mice. In contrast, CD-40, FUS, and control mice tumors showed the presence of more dysfunctional PD-1+ Granzyme B- and non-activated PD-1- Granzyme B T cells. Collectively, these data suggested that adding FUS heating prior to CD-40 tumor treatments protected the T-cells from PD-1 mediated exhaustion, and expanded the population of activated and effector T cells populations rich in IL-2 and IFN-γ; features crucial for systemic immunity and abscopal effects.
The presence of activated innate cells (e.g., macrophages) and Treg can also influence immunotherapy outcomes in patients [Citation49–51]. In particular, tumor-associated macrophages (TAMs) of M1 origin suppress T-cell exhaustion [Citation52,Citation53]. In contrast, M2 macrophages suppress antigen presentation and adaptive immune responses [Citation54]. To dissect the TAM profiles, the M1 and M2 populations in the tumor and spleen tissues were assessed. We noted a significant enhancement of macrophage population of MHCII high M1 phenotype for the FUS40 mice compared to FUS, CD-40, and untreated control. Also, a significant reduction in the population of CD206+ M2 macrophages (∼2-fold; ) in the spleen tissues for FUS40 relative to other treatments was observed. Importantly, the increase of M1 macrophages in the FUS40 tumor was not associated with significant changes in the Treg populations (Figure S2). Tregs infiltrate tumors in response to chemokines secreted in the tumor microenvironments by TAMs (e.g., IL10, a cytokine produced by tumor macrophages) and can inhibit cancer cell cytotoxicity [Citation55–57]. Our data suggest that FUS40 induce the polarization of macrophages without altering the Tregs.
Finally, the systemic administration of anti-CD-40 can damage hepatocytes and impair liver function [Citation32]. A damaged liver is characterized by the release of ALT and AST enzymes from the hepatocytes and decreased albumin producing capacities. We tested if in-situ administration of anti-CD-40 antibody mitigates the adverse liver toxicity outcomes. Data suggested that the serum ALT and AST, and albumin levels were not impacted by CD-40 or FUS, or with FUS40 relative to control (. Thus, the proposed in-situ CD-40 approaches modulated tumor immunity without triggering liver toxicities.
Our study has some limitations. We did not investigate the FUS40 therapeutic effects in a second tumor model. We believe that investigating the local and abscopal effect in tumors that are relatively more immunogenic (e.g. colon) compared to melanoma with FUS40 can shed new lights on the merits of the proposed combinatorial approach for clinical translational. Notably, a recent study in murine Panc02 pancreatic model showed that local CD-40 and radiation (5 Gy) induced infiltrations of T-cells (∼20-fold higher) and improved anti-tumor immunity compared to representative controls [Citation22]. Similarly, another study showed that anti-CD40 antibody and 5 Gy total body irradiation (TBI) increased T‐cell–mediated survival by 100 days in murine B-cell lymphoma [Citation23]. Additionally, a recent phase 1 clinical trials with anti-CD-40 and anti-CTLA-4 therapy in malignant melanoma caused the activation of cytotoxic immune cells and achieved an objective response rate of 27.3% [Citation18]. These promising findings highlight the important role of anti-CD40 in augmenting therapeutic outcomes in the combinatorial regimen, and a need to conduct additional studies in various tumor types with FUS. The second limitation is that we did not notice dramatic differences in tumor growth retardation between anti-CD40 alone and FUS40. We speculate that this is likely due to an insufficient CD40 treatment dosage/frequency or the release of tumor antigens with heating, and the development of adaptive resistance in tumors. Future studies with modulated anti-CD40 dosages, heating conditions, and combinations with other immunotherapies (e.g., checkpoints) can be performed to achieve superior outcomes. Finally, the differences in the immune-activation mechanisms between FUS and tumor irradiation were not compared in our model system. Hypo-fractionated irradiation is known to induce immunogenic death of cancer cells. For example, local irradiation of B16gp melanoma tumors with a single dose of 10 Gy achieved significant retardation of tumor growth by increasing the infiltration of CD45+ leukocytes (2–2.5-folds), enhancement of specific cytotoxic CD8+ T cells, and macrophages [Citation58]. Although promising, the enhanced immune responses with radiation is often inconsistent, and contrastingly some studies also show an increase in the immunosuppressive TGFβ cytokine production, and impaired effector T-cell function [Citation59,Citation60]. Importantly, prior studies conducted in 3LL Lewis lung carcinoma heated to 42–43 °C for 1 h achieved infiltration of DC and T cells in the tumor while also decreasing the regulatory T cells (Treg) and myeloid-derived suppressor cells (MDSC) [Citation42]. Similarly, B16 primary tumors heated to 43 °C for 30 min activated the dendritic and CD8+ T cells in the tumor-draining lymph node (∼1.35-fold) to result in local and systemic tumor growth inhibitions [Citation61]. Furthermore, local heating has been shown to release heat shock protein from cancer cells to enhance sensitization to chemo-, radio- and immune-therapies [Citation62–64]. These promising studies and our current data shows that FUS heating and CD40 can play a crucial role in mitigating the inconsistent immune responses from radiation.
In conclusion, our in vivo data show that FUS40 enhanced the proportion of IL-2, IFN-γ, and Granzyme B rich CD-4+ and CD-8+ T cells and population of M1 macrophages to suppress B16 tumor growth at the treated and untreated site, more so than CD-40 or FUS treatment alone. Studies are currently underway to characterize the role of FUS parameters (hyperthermia vs ablative) and CD-40 treatment sequences to aid the development of a pharmacologic phase 1 clinical trial.
Supplemental Material
Download PDF (540.8 KB)Acknowledgment
Flow cytometry was performed at the Immunopathology Core, Oklahoma Medical Research Foundation and we thank flow cytometry lab members, Dr. Diane Hamilton and Mr. Jacob Bass, for their support. We also thank Mr. Andrew Curtis for processing H and E sections.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Whiteman DC, Green AC, Olsen CM. The growing burden of invasive melanoma: projections of incidence rates and numbers of new cases in six susceptible populations through 2031. J Invest Dermatol. 2016;136(6):1161–1171.
- Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. 2018;8:86–86.
- Taggart D, Andreou T, Scott KJ et al. Anti–PD-1/anti–CTLA-4 efficacy in melanoma brain metastases depends on extracranial disease and augmentation of CD8+ T cell trafficking. Proc Natl Acad Sci USA. 2018;115(7):E1540–E1549.
- Chae YK, Arya A, Iams W, et al. Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC). J Immunother Cancer. 2018;6(1):39.
- Hashimoto M, Kamphorst AO, Im SJ, et al. CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu Rev Med. 2018;69:301–318.
- Hammerich L, Bhardwaj N, Kohrt HE, et al. In situ vaccination for the treatment of cancer. Immunotherapy. 2016;8(3):315–330.
- Hammerich L, Binder A, Brody JD. In situ vaccination: cancer immunotherapy both personalized and off-the-shelf. Mol Oncol. 2015;9(10):1966–1981.
- Clark EA, Yip TC, Ledbettep JA, et al. CDw40 and BLCa-specific monoclonal antibodies detect two distinct molecules which transmit progression signals to human B lymphocytes. Eur J Immunol. 1988;18(3):451–457.
- van Kooten C, Banchereau J. CD40-CD40 ligand. J Leukoc Biol. 2000;67(1):2–17.
- Stout RD, Suttles J, Xu J, et al. Impaired T cell-mediated macrophage activation in CD40 ligand-deficient mice. J Immunol. 1996;156(1):8–11.
- Fransen MF, Sluijter M, Morreau H, et al. Local activation of CD8 T cells and systemic tumor eradication without toxicity via slow release and local delivery of agonistic CD40 antibody. Clin Cancer Res. 2011;17(8):2270–2280.
- Gladue RP, Paradis T, Cole SH, et al. The CD40 agonist antibody CP-870,893 enhances dendritic cell and B-cell activity and promotes anti-tumor efficacy in SCID-hu mice. Cancer Immunol Immunother. 2011;60(7):1009–1017.
- Cella M, Scheidegger D, Palmer-Lehmann K, et al. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J Exp Med. 1996;184(2):747–752.
- Schoenberger SP, Toes RE, van der Voort EI, et al. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393(6684):480–483.
- Rakhmilevich AL, Alderson KL, Sondel PM. T-cell-independent antitumor effects of CD40 ligation. Int Rev Immunol. 2012;31(4):267–278.
- Rakhmilevich AL, Buhtoiarov IN, Malkovsky M, et al. CD40 ligation in vivo can induce T cell independent antitumor effects even against immunogenic tumors. Cancer Immunol Immunother. 2008;57(8):1151–1160.
- Ngiow SF, Young A, Blake SJ, et al. Agonistic CD40 mAb-driven IL12 reverses resistance to anti-PD1 in a T-cell-rich tumor. Cancer Res. 2016;76(21):6266–6277.
- Bajor DL, Mick R, Riese MJ, et al. Long-term outcomes of a phase I study of agonist CD40 antibody and CTLA-4 blockade in patients with metastatic melanoma. Oncoimmunology. 2018;7(10):e1468956.
- Grilley-Olson JE, Curti BD, Smith DC, et al. SEA-CD40, a non-fucosylated CD40 agonist: interim results from a phase 1 study in advanced solid tumors. JCO. 2018;36(15_suppl):3093–3093.
- Hu Z, Yang XY, Liu Y, et al. Investigation of HIFU-induced anti-tumor immunity in a murine tumor model. J Transl Med. 2007;5:34.
- Bandyopadhyay S, Quinn TJ, Scandiuzzi L, et al. Low-intensity focused ultrasound induces reversal of tumor-induced T cell tolerance and prevents immune escape. J Immunol. 2016;196(4):1964–1976.
- Yasmin-Karim S, Bruck PT, Moreau M, et al. Radiation and local anti-CD40 generate an effective in situ vaccine in preclinical models of pancreatic cancer. Front Immunol. 2018;9:2030–2030.
- Honeychurch J, Glennie MJ, Johnson PW, et al. Anti-CD40 monoclonal antibody therapy in combination with irradiation results in a CD8 T-cell-dependent immunity to B-cell lymphoma. Blood. 2003;102(4):1449–1457.
- Fan Y, Kuai R, Xu Y, et al. Immunogenic cell death amplified by co-localized adjuvant delivery for cancer immunotherapy. Nano Lett. 2017;17(12):7387–7393.
- De Palma R, Marigo I, Del Galdo F, et al. Therapeutic effectiveness of recombinant cancer vaccines is associated with a prevalent T-cell receptor alpha usage by melanoma-specific CD8+ T lymphocytes. Cancer Res. 2004;64(21):8068–8076.
- Breshears MA, Eberle R, Ritchey JW. Temporal progression of viral replication and gross and histological lesions in balb/c mice inoculated epidermally with Saimiriine herpesvirus 1 (SaHV-1). J Comparat Pathol. 2005;133(2–3):103–113.
- Dannenmann SR, Thielicke J, Stöckli M, et al. Tumor-associated macrophages subvert T-cell function and correlate with reduced survival in clear cell renal cell carcinoma. Oncoimmunology. 2013;2(3):e23562.
- Maimela NR, Liu S, Zhang Y. Fates of CD8+ T cells in Tumor Microenvironment. Comput Struct Biotechnol J. 2019;17:1–13.
- Hoves S, Ooi C-H, Wolter C, et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J Exp Med. 2018;215(3):859–876.
- Jackaman C, Radley-Crabb HG, Soffe Z, et al. Targeting macrophages rescues age-related immune deficiencies in C57BL/6J geriatric mice. Aging Cell. 2013;12(3):345–357.
- Lin L, Chen Y-S, Yao Y-D, et al. CCL18 from tumor-associated macrophages promotes angiogenesis in breast cancer. Oncotarget. 2015;6(33):34758–34773.
- Medina-Echeverz J, Ma C, Duffy AG, et al. Systemic agonistic anti-CD40 treatment of tumor-bearing mice modulates hepatic myeloid-suppressive cells and causes immune-mediated liver damage. Cancer Immunol Res. 2015;3(5):557–566.
- Diem S, Hasan Ali O, Ackermann CJ, et al. Tumor infiltrating lymphocytes in lymph node metastases of stage III melanoma correspond to response and survival in nine patients treated with ipilimumab at the time of stage IV disease. Cancer Immunol Immunother. 2018;67(1):39–45.
- Liu F, Hu Z, Qiu L, et al. Boosting high-intensity focused ultrasound-induced anti-tumor immunity using a sparse-scan strategy that can more effectively promote dendritic cell maturation. J Transl Med. 2010;8(1):7.
- Huang X, Yuan F, Liang M, et al. M-HIFU inhibits tumor growth, suppresses STAT3 activity and enhances tumor specific immunity in a transplant tumor model of prostate cancer. PLoS One. 2012;7(7):e41632.
- Zhang Y, Deng J, Feng J, et al. Enhancement of antitumor vaccine in ablated hepatocellular carcinoma by high-intensity focused ultrasound. WJG. 2010;16(28):3584–3591.
- de Smet M, Hijnen NM, Langereis S, et al. Magnetic resonance guided high-intensity focused ultrasound mediated hyperthermia improves the intratumoral distribution of temperature-sensitive liposomal Doxorubicin. Invest Radiol. 2013;48(6):395–405.
- Ranjan A, Jacobs GC, Woods DL, et al. Image-guided drug delivery with magnetic resonance guided high intensity focused ultrasound and temperature sensitive liposomes in a rabbit Vx2 tumor model. J Control Rel. 2012;158(3):487–494.
- Manzoor AA, Lindner LH, Landon CD, et al. Overcoming limitations in nanoparticle drug delivery: triggered, intravascular release to improve drug penetration into tumors. Cancer Res. 2012;72(21):5566–5575.
- Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;105(4):256–265.
- Kang J, Demaria S, Formenti S. Current clinical trials testing the combination of immunotherapy with radiotherapy. J Immunother Cancer. 2016;4:51.
- Chen T, Guo J, Han C, et al. Heat shock protein 70, released from heat-stressed tumor cells, initiates antitumor immunity by inducing tumor cell chemokine production and activating dendritic cells via TLR4 pathway. J Immunol. 2009;182(3):1449–1459.
- Chen T, Guo J, Yang M, et al. Chemokine-containing exosomes are released from heat-stressed tumor cells via lipid raft-dependent pathway and act as efficient tumor vaccine. JI. 2011;186(4):2219–2228.
- Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. 2010;129(4):474–481.
- Nelson BH. IL-2, regulatory T cells, and tolerance. J Immunol. 2004;172(7):3983–3988.
- Liao W, Lin J-X, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013;38(1):13–25.
- Mellor-Heineke S, Villanueva J, Jordan MB, et al. Elevated granzyme B in cytotoxic lymphocytes is a signature of immune activation in hemophagocytic lymphohistiocytosis. Front Immunol. 2013;4:72.
- Chikuma S, Terawaki S, Hayashi T, et al. PD-1-mediated suppression of IL-2 production induces CD8+ T cell anergy in vivo. J Immunol. 2009;182(11):6682–6689.
- Ji R-R, Chasalow SD, Wang L, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012; 61(7):1019–1031.
- Gajewski TF, Louahed J, Brichard VG. Gene signature in melanoma associated with clinical activity: a potential clue to unlock cancer immunotherapy. Cancer J. 2010;16(4):399–403.
- Hamid O, Schmidt H, Nissan A, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. 2011;9(1):204.
- Wiehagen KR, Girgis NM, Yamada DH, et al. Combination of CD40 agonism and CSF-1R blockade reconditions tumor-associated macrophages and drives potent antitumor immunity. Cancer Immunol Res. 2017;5(12):1109–1121.
- Baer C, Squadrito ML, Laoui D, et al. Suppression of microRNA activity amplifies IFN-γ-induced macrophage activation and promotes anti-tumour immunity. Nat Cell Biol. 2016;18(7):790.
- Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51.
- Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors in cancer therapy: a focus on T-regulatory cells. Immunol Cell Biol. 2018;96(1):21–33.
- Sakai K, Maeda S, Yamada Y, et al. Association of tumour-infiltrating regulatory T cells with adverse outcomes in dogs with malignant tumours. Vet Comp Oncol. 2018;16(3):330.
- Zhu Q, Wu X, Wu Y, et al. Interaction between Treg cells and tumor-associated macrophages in the tumor microenvironment of epithelial ovarian cancer. Oncol Rep. 2016;36(6):3472–3478.
- Gupta A, Probst HC, Vuong V, et al. Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J Immunol. 2012;189(2):558.
- Golden EB, Frances D, Pellicciotta I, et al. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology. 2014;3(4):e28518.
- Gameiro SR, Jammeh ML, Wattenberg MM, et al. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget. 2014;5(2):403–416.
- Toraya-Brown S, Sheen MR, Zhang P, et al. Local hyperthermia treatment of tumors induces CD8(+) T cell-mediated resistance against distal and secondary tumors. Nanomedicine. 2014;10(6):1273–1285.
- Behrouzkia Z, Joveini Z, Keshavarzi B, et al. Hyperthermia: how can it be used? Oman Med J. 2016;31(2):89–97.
- Rao W, Deng ZS, Liu J. A review of hyperthermia combined with radiotherapy/chemotherapy on malignant tumors. Crit Rev Biomed Eng. 2010;38(1):101–116.
- Datta NR, Ordóñez SG, Gaipl US, et al. Local hyperthermia combined with radiotherapy and-/or chemotherapy: recent advances and promises for the future. Cancer Treat Rev. 2015;41(9):742–753.