
Abstract
Objectives
Angiogenesis occurs during tumor progression of hepatocellular carcinoma (HCC) after insufficient radiofrequency ablation (RFA). Arsenic trioxide (ATO) shows promising therapeutic potential in advanced HCC. Whether ATO regulates angiogenesis and can be used to prevent tumor progression in HCC after insufficient RFA is still unknown.
Methods
Insufficient RFA was simulated using a water bath. MTT assay and tube formation assay were used to evaluate the effects of ATO on viability and proangiogenic abilities of SMMC7721 and HepG2 cells after insufficient RFA in vitro. The molecular changes with the treatment of ATO were evaluated through Western blot. An ectopic nude mice model was used to evaluate the effect of ATO on the tumor of SMMC7721 cells in vivo after insufficient RFA.
Results
In this study, HepG2 and SMMC7721 cells after insufficient RFA (named HepG2-H and SMMC7721-H, respectively) showed higher proliferation than the untreated cells and promoted tube formation of endothelial cells in a paracrine manner. ATO eliminated the difference in proliferation between untreated and RFA-treated cells and suppressed angiogenesis induced by HCC cells after insufficient RFA through the Ang-1 (angiopoietin-1)/Ang-2 (angiopoietin-2)/Tie2 pathway. Hif-1α overexpression abolished the inhibitory effect of ATO on angiogenesis in HCC after insufficient RFA. ATO inhibited tumor growth and angiogenesis in HCC after insufficient RFA.
Conclusions
Our results demonstrate that ATO blocks the paracrine signaling of Ang-1 and Ang-2 by inhibiting p-Akt/Hif-1α and further suppresses the angiogenesis of HCC after insufficient RFA.
Introduction
Hepatocellular carcinoma (HCC) is the most common primary liver neoplasm and the third leading cause of cancer-related mortality worldwide [Citation1]. Surgical resection, liver transplantation and radiofrequency ablation (RFA) are the standard first-line approaches to treat early-stage HCC [Citation2,Citation3].
However, local recurrences post-RFA are higher than post-surgical resection, and insufficient RFA may cause enhanced growth and vascular invasion [Citation4,Citation5]. Angiogenesis occurs during tumor progression in HCC after insufficient RFA. Our previous study demonstrated enhanced angiogenesis in HCC after insufficient RFA and the involvement of Hif-1α/VEGFA in this process [Citation6]. Tumor endothelial cells showed increased angiogenesis after insufficient RFA and facilitated the invasiveness of residual HCC [Citation7]. Moreover, ATPase inhibitory factor 1 was involved in the angiogenesis of HCC after insufficient RFA [Citation8]. Insufficient RFA promoted HCC cell proliferation by stimulating VEGF overexpression, [Citation9] and the growth of non-small cell lung cancer cells through PI3K/Akt/Hif-1α [Citation10], which was involved in tumor angiogenesis. HCC cells after insufficient RFA promote angiogenesis via VEGFA; however, the therapeutic effects of anti-VEGFA antibody (bevacizumab) are limited in HCC. Therefore, more targeted therapies are urgently required. Angiopoietin-1 (Ang-1) and angiopoietin-2 (Ang-2) are secretory proteins, which are important angiogenic factors and regulate tumor angiogenesis [Citation11,Citation12]. However, whether Ang-1 and Ang-2 are involved in the angiogenesis of HCC after insufficient RFA is largely unexplored. In addition, interaction between HCC and endothelial cells during angiogenesis in HCC remains unclear.
Arsenic trioxide (ATO) has been approved as the first-line treatment for acute promyelocytic leukemia by the Food and Drug Administration [Citation13]. ATO shows promising therapeutic potential against advanced HCC by reducing hepatic metastasis and prolonging survival. ATO can suppress epithelial–mesenchymal transition, cancer stem cells, and angiogenesis, induce apoptosis and increase drug sensitivity in HCC [Citation14–18]. Meanwhile, ATO can improve tumor destruction in RFA [Citation19–21]. Nevertheless, the mechanism by which ATO is involved in angiogenesis and whether ATO can be used to prevent tumor progression in HCC after insufficient RFA are still unknown.
In this study, we hypothesized that the high expression of p-AKT/Hif-1α in HCC after insufficient RFA regulates paracrine Ang-1 and Ang-2, which promote tube formation of endothelial cells. We evaluated the role of ATO and the underlying mechanism in suppressing angiogenesis in HCC after insufficient RFA. Our study indicates suitable targets to develop novel and effective therapies to prevent angiogenesis in HCC following insufficient RFA.
Materials and methods
Ethics statement
All animal experiments were approved by the Animal Care Committee of Capital Medical University (Beijing, China) and performed in accordance with the institutional guidelines.
Cell culture
Human HCC cell lines, SMMC7721 and HepG2, were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). Cells were maintained in high-glucose Dulbecco’s modified Eagle medium (DMEM; Gibco, UK) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin (Life Technologies, Cergy Pontoise, France) in a humidified atmosphere of 5% CO2 at 37 °C.
Human umbilical vein endothelial cells (HUVECs) were obtained from Sciencell (San Diego, CA, USA) and cultured in Endothelial Cell Medium supplemented with 10% FBS, 1% endothelial cell growth supplement, 100 U/mL penicillin and 100 μg/mL streptomycin (ECM; Gibco, UK) in a humidified incubator at 3 °C and 5% CO2.
Reagents and antibodies
ATO was purchased from Sigma-Aldrich (St. Louis, MO, USA), its stock solution (10 mmol/L) was prepared in phosphate-buffered saline (PBS) and kept at 4 °C. The Hif-1α inhibitor, YC-1 [3-(50-hydroxy methyl-20-furyl)-1-benzylindazole], and MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] were purchased from Sigma-Aldrich (St. Louis, MO, USA). Anti-p-Akt (#4060) and anti-T-Akt (#4685) antibodies were purchased from Cell Signaling Technology (Beverly, CA, USA). Anti- Ang-1 (AF923), Ang-2 (MAB7186; MAB098) and anti-Tie2 (AF313) antibodies were purchased from R&D Systems (Minneapolis, MN, USA). Anti-Hif-1α (ab228649) and anti-CD31 (ab187377) antibodies were purchased from Abcam (Cambridge, TX, USA). Anti-β-actin antibody (sc-8342) was obtained from Santa Cruz Biotechnology (Dallas, TX, USA).
Heat treatment
Briefly, HCC cells were seeded into 6-well plates (5 × 104 cells/well). After 24 h, the plates were sealed and submerged in a water bath set to 37 or 47 °C for 5 min. The cells were allowed to recover, and when the surviving populations reached 80% confluence, they were propagated into 6-well plates and exposed to the above heat treatment again for 10 min. The process was repeated, and cells were sequentially exposed to the above heat treatment for 15, 20 and 25 min. Cells surviving the treatment at 47 °C for 25 min were designated as SMMC7721-H and HepG2-H cells. Cells treated at 37 °C were SMMC7721 and HepG2 cells.
MTT assay
Cells preincubated in 96-well plates at 3 × 103/well density were treated with different ATO concentrations (0, 1, 2 or 5 µmol/L) and cell viability was assessed after 24, 48 and 72 h. The medium was discarded after ATO treatment, and the cells were washed with PBS. MTT solution was added to each well at a final concentration of 0.5 mg/mL and incubated for 4 h. Formazan crystals, resulting from MTT reduction, were dissolved in 150 μL of dimethyl sulfoxide per well, and the absorbance was measured at 570 nm using an automated ELISA plate reader.
Collection of the conditioned medium (CM)
HCC cells were incubated in 6-well plates in DMEM supplemented with 10% FBS for 24 h. The cells were then treated with ATO for 24 h after starvation for 8 h. The medium was replaced with free ECM after washing the cells with PBS. After 4 h, CM was collected and centrifuged at 1000 × g for 20 min, and the supernatant was stored at −80 °C.
ELISA
Ang-1 and Ang-2 levels in CM were quantified using an ELISA kit (R&D Systems, MN, USA) according to the manufacturer’s instructions.
Tube formation assay
HUVECs were trypsinized and seeded at a density of 1.5 × 104 cells/well in a 96-well plate on Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) that was polymerized for 30 min at 37 °C. The cells were then incubated in CM for 4 h. Capillary morphogenesis was evaluated using an inverted microscope (Olympus, Tokyo, Japan). Tube formation was assessed by counting tube rings from five random fields at 100× magnification. The relevant effect of CM was normalized with respect to that of the total cellular proteins.
siRNA transfection
The siRNA sequences used are as follows. Ang-1 siRNA: GGAGAAGATATAACCGGAT; Ang-2 siRNA: GGACAAACCTGTTGAACCAAA; and Tie2 siRNA: GCUUCUAUACAAACCCGUUTT. They were synthesized by Shanghai GenePharma Co. (Shanghai, China). HCC cells were plated in 6-well plates and allowed to grow to sub-confluency. Cells were transiently transfected with siRNAs using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) in Opti-MEM medium (Gibco) for 12 h and used for further experiments.
Hif-1α overexpression in HCC cells
Human HCC cells in 6-well plates were transfected with pcDNA3.1(-)-Hif-1α plasmid containing Hif-1α in culture medium using Lipofectamine 2000 (Invitrogen, Shanghai, China) according to the manufacturer’s instructions. Briefly, HCC cells were seeded onto 6-well culture plates at a density of 5 × 105 cells/well and grown to 60–70% confluence in complete media. Then 2.5 µg pcDNA3.1(-)-Hif-1α (overexpression, Over) or pcDNA3.1(+) (negative control, NC) was mixed with 10 µL Lipofectamine 2000 and diluted in 2000 µL serum-free DMEM. After 5 min of incubation, DNA-liposome complexes were added to the cells and incubated for 6 h at 37 °C and 5% CO2. Complete DMEM containing 10% FBS was added to the cells and incubated overnight, and the medium was replaced with complete DMEM. Western blot was performed to analyze the transfection efficiency by assessing Hif-1α levels.
Western blot analysis
After drug treatment, cells were lysed in lysis buffer (150 mM NaCl, 50 mM Tris-HCl, pH 8.0, 0.1% SDS, 1% Triton X-100) containing protease and phosphatase inhibitors. Protein concentrations in cell lysates were measured using a bicinchoninic acid protein assay kit (KeyGEN BioTECH, Nanjing, China). Equal amounts of protein (50 μg) were separated using SDS-PAGE and transferred to nitrocellulose membranes (Millipore, Billerica, USA). The membranes were blocked with 5% nonfat dry milk in tris-buffered saline containing 0.1% Tween 20 for 2 h, and subsequently probed with respective primary antibodies overnight at 4 °C. The membranes were then incubated with HRP-conjugated secondary antibodies for 1.5 h at room temperature. Blots were visualized with an ECL detection kit (Pierce, USA), acquired using Quantity One 1-D Analysis Software (Bio-Rad, Hercules, CA, USA) and analyzed using ImageJ software.
Xenograft model
BALB/C nude mice (male, four week old) were purchased from Vital River (Beijing, China) and maintained in a barrier facility under pathogen-free conditions. The mice were fed an autoclaved laboratory rodent diet. SMMC7721 and SMMC7721-H cells (5 × 106 cells each) were suspended in 200 μL of serum-free DMEM and Matrigel (1:1) and subcutaneously injected into the upper right flank region of 20 nude mice. Subsequently, mice were intraperitoneally injected with ATO (1 mg.kg−1.d−1) or PBS (control) once a day for four weeks. Tumor size was measured using a caliper every other day. Tumor volume was calculated as follows: (L × S2)/2, where L and S are the longest and shortest diameters in centimeter. At the end of the experiments, the mice were euthanized, and tumor tissues were removed and fixed in 4% paraformaldehyde for histological examination and immunohistochemical staining.
Immunohistochemistry analysis
In brief, after dewaxing, rehydration, and antigen retrieval, permeabilized sections (6 μm) were blocked with 5% goat serum for 60 min at 37 °C, and incubated overnight with respective primary antibodies. They were subsequently incubated with HRP-conjugated secondary antibodies for 30 min at 37 °C, and then developed with 3,3′-diaminobenzidine. The slides were counterstained with hematoxylin, mounted, and examined using a Nikon Eclipse Ti microscope under a 200× objective. Images were analyzed using Image-Pro Plus 6.0.
Statistical analysis
Data are represented as the mean ± standard deviation (SD). The IC50 values were deduced from log-normalized data using GraphPad Prism. Data were analyzed using analysis of variance. Results with p < 0.05 were considered statistically significant. GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA) was used for statistical analyses.
Results
ATO at low concentrations does not inhibit the proliferation of HCC cells after insufficient RFA
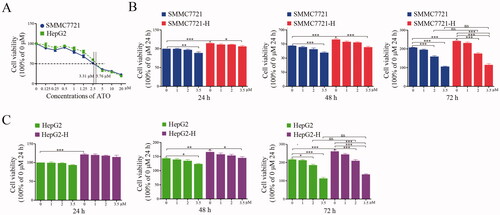
To assess the inhibitory effects of ATO on HCC cells, we calculated the IC50 of ATO using MTT assay. The IC50 values of ATO in HepG2 and SMMC7721 cells were 3.76 and 3.31 μmol/L, respectively (). Therefore, we selected 3.5 μmol/L as the maximum ATO concentration for further experiments. We examined the effects of ATO on HCC cells after insufficient RFA treatment. HepG2-H and SMMC7721-H cells showed 20.8% and 16.9% increases in proliferation at 72 h compared with those of HepG2 and SMMC7721 cells, respectively ()). ATO at 2 and 3.5 μmol/L doses significantly suppressed the proliferation of HCC cells ()). Furthermore, 3.5 μmol/L ATO eliminated the difference in proliferation between RFA-treated and untreated HCC cells ()). However, 1 μmol/L ATO did not show this effect ()). In other words, HepG2-H or SMMC7721-H cells showed no more sensitivity to ATO at the concentrations of 1 μmol/L compared with HepG2 or SMMC7721 cells respectively.
Figure 1. ATO at low concentrations does not inhibit the proliferation of HCC cells after insufficient RFA. HCC cells were treated with ATO and MTT assay was performed to assess the inhibitory effects on cell proliferation. (A) SMMC7721or HepG2 was treated with ATO for 72 h and IC50 was calculated. (B) SMMC7721 and SMMC7721-H cells were treated with ATO at 1, 2, and 3.5 μM concentrations. The differences in inhibitory effects of ATO on SMMC7721 and SMMC7721-H cells are shown. (C) HepG2 and HepG2-H cells were treated with ATO at 1, 2, and 3.5 μM concentrations. The differences in inhibitory effects of ATO on HepG2 and HepG2-H cells are shown. At least three independent experiments were performed. ns, no significance; *p<0.05; **p<0.01; and ***p<0.001.
HCC cells after insufficient RFA promote tube formation of endothelial cells in a paracrine manner, which is suppressed by ATO
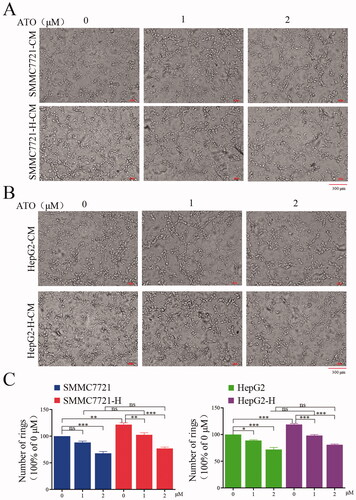
We assessed whether 1 and 2 μmol/L ATO inhibits tumor angiogenesis in a paracrine manner. We collected CM from HCC cells with and without ATO treatment. The effect of CM on endothelial cells was assessed by analyzing tube formation. Endothelial cells showed an enhanced capacity of tube formation after treatment with CM from SMMC7721-H or HepG2-H cells compared with that from SMMC7721 or HepG2 cells (). ATO treatment impaired CM-induced tube formation (). Meanwhile, ATO treatment abolished the difference in promoting tube formation between CM from RFA-treated and untreated HCC cells ().
Figure 2. HCC cells after insufficient RFA promote tube formation of endothelial cells by paracrine manner, and ATO suppresses the process. CM was collected from HCC cells or HCC cells after insufficient RFA with or without ATO treatment and used to treat endothelial cells. CM from (A) SMMC7721 or SMMC7721-H cells and (B) HepG2 or HepG2-H cells, with or without ATO treatment was used to treat endothelial cells and tube formation was examined. (C) Statistical results of (A) and (B) are shown. At least three independent experiments were performed. ns, no significance; *p<0.05; **p<0.01; and ***p<0.001.
ATO inhibits angiogenesis induced by HCC cells after insufficient RFA through the Ang1/Ang2/Tie2 pathway
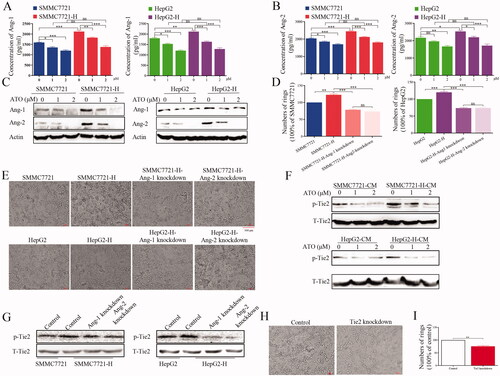
We examined the factors secreted in CM from HCC cells. Higher levels of Ang-1 and Ang-2 were found in CM from RFA-treated than from untreated HCC cells; however, ATO decreased the levels of Ang-1 and Ang-2 in CM (). Meanwhile, 1 and 2 μmol/L ATO abolished the difference in Ang-1 and Ang-2 levels in CM between RFA-treated and untreated HCC cells (). Furthermore, the expression of Ang-1 and Ang-2 in RFA-treated HCC cells was higher than that in untreated cells, and ATO inhibited the expression of Ang-1 and Ang-2 (, Figure S1(a)). After Ang-1 or Ang-2 knockdown (Figure S2), the ability of CM to promote angiogenesis was impaired, which indicated that Ang-1 and Ang-2 participated in enhancing angiogenesis ()). p-Tie2 expression was upregulated after treatment with CM from RFA-treated HCC cells compared with that from untreated cells; ATO inhibited p-Tie2 expression (, Figure S1(b)). Meanwhile, after Ang-1 or Ang-2 knockdown, the effect of CM from HCC cells on p-Tie2 expression was eliminated (, Figure S1(c)). We also used Tie2 siRNA to knockdown Tie2 expression in endothelial cells (Figure S2). The ability of CM to promote angiogenesis was impaired following Tie2 knockdown ()). These results indicate that ATO suppresses angiogenesis through the Ang-1/Ang-2/Tie2 pathway.
Figure 3. ATO inhibits angiogenesis induced by HCC cells after insufficient RFA through the Ang-1/Ang-2/Tie2 pathway. (A, B) The concentration of Ang-1 and Ang-2 in conditioned media from SMMC7721 and SMMC7721-H, or HepG2 and HepG2-H cells, with or without ATO treatment was measured. (C) The expression of Ang-1 or Ang-2 in HCC cells and HCC cells after insufficient RFA was examined using Western blot. (D, E) Ang-1 or Ang-2 was knocked down in SMMC7721-H or HepG2-H cells, CM was collected, and tube formation was assessed. (F) CM from untreated or RFA-treated HCC cells with or without ATO treatment was used to treat endothelial cells, and p-Tie2 expression was analyzed. (G) Ang-1 or Ang-2 was knocked down, CM was used to treat endothelial cells, and p-Tie2 expression was analyzed. (H, I) Tie2 was knocked down in endothelial cells, and the effect of CM from HCC cells on tube formation was examined. At least three independent experiments were performed. *p<0.05; **p<0.01; ***p<0.001; and ns: no significance.
Hif-1α overexpression abolishes the inhibiting effect of ATO on angiogenesis after insufficient RFA
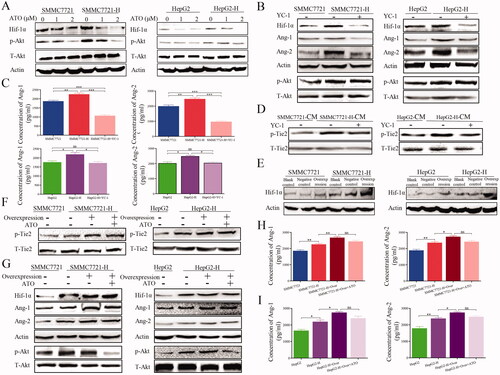
We explored whether Hif-1α was involved ATO-induced inhibition of angiogenesis in HCC after insufficient RFA. ATO suppressed the expression of Hif-1α and p-Akt in HCC cells ( and Figure S3(a)), indicating that p-Akt/Hif-1α may be involved in angiogenesis here. Therefore, we inhibited Hif-1α using YC-1 in HCC cells, and found that YC-1 inhibited the increased expression of Hif-1α, Ang-1 and Ang-2 in HepG2-H or SMMC7721-H cells ( and Figure S3(b)). YC-1 suppressed the levels of Ang-1 and Ang-2 in HepG2-H and SMMC7721-H cells () and downregulated p-Tie2 expression in HCC cells after insufficient RFA ( and Figure S3(C)). Next, we used lentivirus to overexpress Hif-1α in HCC cells ( and Figure S3(d)). p-Tie2 expression increased following Hif-1α overexpression, and the effect of ATO was impaired ( and Figure S3(e)). After Hif-1α overexpression, the levels of Ang-1, Ang-2 and p-Akt were also upregulated in HCC cells ( and Figure S3(f)), and the inhibitory effect of ATO was impaired (, Figure S3(f)). These results suggest that ATO suppresses Ang-1 and Ang-2 via the Hif-1α/p-Akt signaling pathway.
Figure 4. Hif-1α overexpression abolishes the inhibitory effect of ATO on angiogenesis after insufficient RFA. (A) HCC cells were treated with ATO and the expression of Hif-1α and p-Akt was analyzed. (B) HCC cells were treated with YC-1 and the expression of Hif-1α, p-Akt, Ang-1, and Ang-2 was analyzed. (C) The concentrations of Ang-1 and Ang-2 in CM after YC-1 treatment were measured in HCC cells after insufficient RFA. (D) HCC cells were treated with YC-1 and p-Tie2 expression was analyzed. (E) Western blot was used to validate the transfection efficacy of plasmid containing Hif-1α. (F) After Hif-1α overexpression, HCC cells were treated with ATO, CM was collected and used to treat endothelial cells, and p-Tie2 expression was analyzed. (G) HCC cells with Hif-1α overexpression were treated with ATO and the expression of p-Akt, Hif-1α, Ang-1, and Ang-2 was analyzed. (H, I) HCC cells with Hif-1α overexpression were treated with ATO and the concentrations of Ang-1 and Ang-2 in CM were determined. At least three independent experiments were performed. BC: Blank control; NC: Negative control; and Over: Overexpression. *p<0.05; **p<0.01; ***p<0.001; and ns: no significance.
ATO restrains tumor growth and angiogenesis of HCC after insufficient RFA
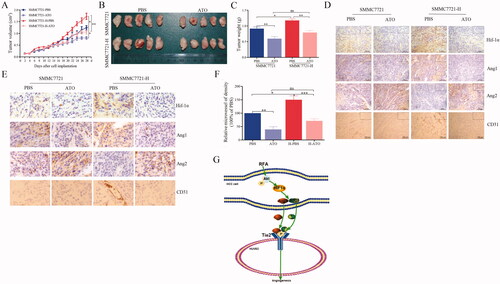
We evaluated the effects of ATO on HCC cells in vivo. The experimental mice were divided into four groups: (1) SMMC7721 + PBS, (2) SMMC7721-H + PBS, (3) SMMC7721 + ATO, and (4) SMMC7721-H + ATO. Group (2) showed rapid tumor growth compared with group (1). The mice were administered 1 mg/kg ATO (conversion from human dose) or PBS by intraperitoneal injection every day and examined for tumor growth. ATO significantly inhibited tumor growth in groups (3) and (4) (). In addition, ATO ameliorated the differences in tumor growth between SMMC7721 and SMMC7721-H cells (). Higher expression of Ang-1, Ang-2 and Hif-1α was observed in Group (2) than in Group (1) ()). Group (2) showed higher microvessel density than Group (1) (). ATO reduced the expression of Ang-1, Ang-2 and Hif-1α, and microvessel density in SMMC7721 and SMMC7721-H tumors ( and Figure S4). The schematic shows that ATO inhibits tube formation by endothelial cells in a paracrine manner in HCC after insufficient RFA (). No apparent changes were observed in body weight, heart, liver, spleen, lung, kidney, and liver and renal function in mice in tumor xenograft experiment (Figure S5).
Figure 5. ATO restrains tumor growth and angiogenesis in HCC after insufficient RFA. (A) SMMC7721 and SMMC7721-H cells were subcutaneously inoculated into nude mice, which were then treated with PBS or ATO. After four weeks, the animals were sacrificed. Tumor size was measured with a caliper after every other day. (B) Images representing tumor volume at the end of experiments, n = 5 per group. (C) Tumor weight was measured at the end of the experiment. (D) Tumor sections were stained for the detection of Hif-1α, Ang-1, Ang-2, or CD31. Representative images of immunohistochemical analysis. (E) Representative images with high magnification. (F) Quantification of CD31. (G) Schematic shows the effect of ATO on endothelial cells and the underlying mechanisms, in HCC after insufficient RFA. At least three independent experiments were performed. *p<0.05; **p<0.01; and ns: no significance.
Discussion
ATO is an FDA-approved drug to treat acute promyelocytic leukemia and has great potential in treating solid tumors. However, the mechanism through which ATO suppresses angiogenesis in HCC remains unclear. In the present study, ATO at low concentrations blocked the paracrine signaling of Ang-1 and Ang-2 by inhibiting p-Akt/Hif-1α and suppressed angiogenesis of HCC after insufficient RFA.
ATO exerts anti-proliferative effects against solid tumors [Citation22]; however, the chemotherapeutic effect in solid tumors is achieved at high doses [Citation23], which is not clinically achievable without the risk of various side effects, including skin disorders, internal cancers, cardiovascular disease and reproductive disorders [Citation24,Citation25]. Furthermore, most patients with HCC cannot undergo systemic chemotherapy because of hepatic insufficiency or medical comorbidities. Therefore, ATO should be applied at a low dose owing to its poor biosafety and narrow drug-safety window, and the effects and mechanism of action at low doses of ATO should be explored. Administration of 10 mg/d ATO in acute promyelocytic leukemia patients resulted in a rapid increase in plasma arsenic levels that reached the peak with a mean Cpmax of 6.85 μmol/L. However, plasma arsenic levels rapidly dropped to 2 μmol/L after 10 h of ATO administration. In the present study, the IC50 values of HepG2 and SMMC7721 cells were 3.31 and 3.76 μmol/L; therefore, 1, 2, and 3.5 μmol/L ATO doses were selected for further experiments. We speculated that ATO at low concentrations exerts an inhibitory effect on HCC after insufficient RFA.
Abnormal angiogenesis is an important characteristic of HCC, which provides nutrient and oxygen to tumors and assists tumor cells in spreading through the bloodstream to distant organs [Citation26]. Our previous study also demonstrated that angiogenesis was enhanced in HCC after insufficient RFA, and Hif-1α and VEGFA were involved in this process [Citation6]. Tumor endothelial cells show increased angiogenesis after insufficient RFA and facilitate the invasiveness of residual hepatocellular carcinoma [Citation7]. Therefore, we tested whether ATO suppresses angiogenesis in HCC after insufficient RFA. Our results showed that CM from HCC cells after insufficient RFA promoted tube formation by HUVECs, and ATO at 1 and 2 μmol/L doses suppressed tube formation. The difference in tube formation between CM from RFA-treated and untreated HCC cells was eliminated after ATO treatment. Therefore, ATO at low concentrations may suppress HCC progression by inhibiting angiogenesis.
Angiopoietins are a family of secreted factors comprising Ang-1, Ang-2, angiopoietin-3, and angiopoietin-4 [Citation27]. Ang-1 and Ang-2 are important factors that regulate angiogenesis through the TEK tyrosine kinase endothelial receptor and have been identified as potential prognostic biomarkers of HCC [Citation28,Citation29]. Our previous study showed that Hif-1α/VEGFA mediates angiogenesis in HCC after insufficient RFA [Citation6]. Therefore, we investigated whether other signaling pathways are involved in this process. We found that the levels of Ang-1 and Ang-2 were upregulated in HCC cells after insufficient RFA, and knockdown of Ang-1 and Ang-2 or Tie2 attenuated the effect of CM from HCC cells after insufficient RFA promoting tube formation of endothelial cells. ATO exerts its anti-tumor effect by inhibiting the STAT3, PI3K/AKT and MAPK signaling pathways. Therefore, we examined whether ATO suppresses Ang-1 and Ang-2 expression. In the present study, ATO decreased the expression of Ang-1 and Ang-2. Therefore, ATO suppresses angiogenesis of HCC after insufficient RFA through downregulation of Ang-1 and Ang-2.
Hif-1α is a well-defined hypoxia-responsive factor that activates diverse pathways to regulate cellular metabolism, angiogenesis, proliferation and drug resistance [Citation30]. We found that ATO inhibited Hif-1α expression in HCC cells after insufficient RFA. Hif-1α overexpression abolished the inhibitory effect of ATO on Ang-1 and Ang-2. Furthermore, Hif-1α overexpression attenuated the effect of ATO on the inhibition of tube formation in HUVECs. These results suggest that ATO suppresses the expression of Ang-1 and Ang-2 via Hif-1α.
In conclusion, our study showed that high expression of p-Akt/Hif-1α in HCC after insufficient RFA regulates paracrine Ang-1 and Ang-2 secretion, which promote tube formation of endothelial cells. Furthermore, ATO, at relatively low concentrations, inhibits angiogenesis in HCC by blocking paracrine Ang-1 and Ang-2.
Acknowledgements
The authors thank Taylor and Francis editing service (http://www.tandfeditingservices.com) for editing the English text of a draft of this manuscript.
Disclosure statement
The authors report there are no competing interests to declare.
Data availability statement
The datasets used and analyzed during the study are available from the corresponding author upon reasonable request.
References
- McGlynn KA, Petrick JL, El-Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology. 2021;73 (Suppl 1):4–13.
- Kloeckner R, Galle PR, Bruix J. Local and regional therapies for hepatocellular carcinoma. Hepatology. 2021;73(S1):137–149.
- Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6.
- Shin SW, Ahn KS, Kim SW, et al. Liver resection versus local ablation therapies for hepatocellular carcinoma within the Milan criteria: a systematic review and meta-analysis. Ann Surg. 2021;273(4):656–666.
- Lee S, Kang TW, Song KD, et al. Effect of microvascular invasion risk on early recurrence of hepatocellular carcinoma after surgery and radiofrequency ablation. Ann Surg. 2021;273(3):564–571.
- Kong J, Kong J, Pan B, et al. Insufficient radiofrequency ablation promotes angiogenesis of residual hepatocellular carcinoma via HIF-1alpha/VEGFA. PLoS One. 2012;7(5):e37266.
- Kong J, Kong L, Kong J, et al. After insufficient radiofrequency ablation, tumor-associated endothelial cells exhibit enhanced angiogenesis and promote invasiveness of residual hepatocellular carcinoma. J Transl Med. 2012;10(1):230.
- Kong J, Yao C, Ding X, et al. ATPase inhibitory factor 1 promotes hepatocellular carcinoma progression after insufficient radiofrequency ablation, and attenuates cell sensitivity to sorafenib therapy. Front Oncol. 2020;10:1080.
- Tan L, Chen S, Wei G, et al. Sublethal heat treatment of hepatocellular carcinoma promotes intrahepatic metastasis and stemness in a VEGFR1-dependent manner. Cancer Lett. 2019;460:29–40.
- Wan J, Wu W, Chen Y, et al. Insufficient radiofrequency ablation promotes the growth of non-small cell lung cancer cells through PI3K/akt/HIF-1alpha signals. Acta Biochim Biophys Sin (Shanghai). 2016;48(4):371–377.
- Fagiani E, Christofori G. Angiopoietins in angiogenesis. Cancer Lett. 2013;328(1):18–26.
- Reiss Y. Angiopoietins. Recent Results Cancer Res. 2010;180:3–13.
- Sanz MA, Fenaux P, Tallman MS, et al. Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood. 2019;133(15):1630–1643.
- Huang Y, Zhou B, Luo H, et al. ZnAs@SiO2 nanoparticles as a potential anti-tumor drug for targeting stemness and epithelial-mesenchymal transition in hepatocellular carcinoma via SHP-1/JAK2/STAT3 signaling. Theranostics. 2019;9(15):4391–4408.
- Wang HY, Zhang B, Zhou JN, et al. Arsenic trioxide inhibits liver cancer stem cells and metastasis by targeting SRF/MCM7 complex. Cell Death Dis. 2019;10(6):453.
- Jiang F, Wang X, Liu Q, et al. Inhibition of TGF-β/SMAD3/NF-κB signaling by microRNA-491 is involved in arsenic trioxide-induced anti-angiogenesis in hepatocellular carcinoma cells. Toxicol Lett. 2014;231(1):55–61.
- Cai X, Yu L, Chen Z, et al. Arsenic trioxide-induced upregulation of miR-1294 suppresses tumor growth in hepatocellular carcinoma by targeting TEAD1 and PIM1. Cancer Biomark. 2020;28(2):221–230.
- Zhang X, Hu B, Sun YF, et al. Arsenic trioxide induces differentiation of cancer stem cells in hepatocellular carcinoma through inhibition of LIF/JAK1/STAT3 and NF-kB signaling pathways synergistically. Clin Transl Med. 2021;11(2):e335.
- Hines-Peralta A, Sukhatme V, Regan M, et al. Improved tumor destruction with arsenic trioxide and radiofrequency ablation in three animal models. Radiology. 2006;240(1):82–89.
- Seong NJ, Yoon CJ, Kang SG, et al. Effects of arsenic trioxide on radiofrequency ablation of VX2 liver tumor: intraarterial versus intravenous administration. Korean J Radiol. 2012;13(2):195–201.
- Horkan C, Ahmed M, Liu Z, et al. Radiofrequency ablation: Effect of pharmacologic modulation of hepatic and renal blood flow on coagulation diameter in a VX2 tumor model. J Vasc Interv Radiol. 2004;15(3):269–274.
- Sönksen M, Kerl K, Bunzen H. Current status and future prospects of nanomedicine for arsenic trioxide delivery to solid tumors. Med Res Rev. 2022;42(1):374–398.
- Liu B, Pan S, Dong X, et al. Opposing effects of arsenic trioxide on hepatocellular carcinomas in mice. Cancer Sci. 2006;97(7):675–681.
- Haybar H, Shahrabi S, Rezaeeyan H, et al. Strategies to inhibit arsenic trioxide-induced cardiotoxicity in acute promyelocytic leukemia. J Cell Physiol. 2019;234(9):14500–14506.
- Kulik-Kupka K, Koszowska A, Bronczyk-Puzon A, et al. [Arsenic - Poison or medicine?]. Med Pr. 2016;67(1):89–96.
- Morse MA, Sun W, Kim R, et al. The role of angiogenesis in hepatocellular carcinoma. Clin Cancer Res. 2019;25(3):912–920.
- Yu X, Ye F. Role of angiopoietins in development of cancer and neoplasia associated with viral infection. Cells. 2020;9(2):457.
- Lin N, Meng L, Lin J, et al. Activated hepatic stellate cells promote angiogenesis in hepatocellular carcinoma by secreting angiopoietin-1. J Cell Biochem. 2020;121(2):1441–1451.
- Xie JY, Wei JX, Lv LH, et al. Angiopoietin-2 induces angiogenesis via exosomes in human hepatocellular carcinoma. Cell Commun Signal. 2020;18(1):46.
- Paredes F, Williams HC, San Martin A. Metabolic adaptation in hypoxia and cancer. Cancer Lett. 2021;502:133–142.