
Abstract
Primary objective: Excessive accumulation of amyloid beta (Aβ) and tau have been observed in older individuals with chronic neurological symptoms related to a traumatic brain injury (TBI), yet little is known about the possible role of Aβ in younger active duty service members following a TBI. The purpose of the study was to determine if Aβ 40 or 42 related to sustaining a TBI or to chronic neurological symptoms in a young cohort of military personnel.
Research design: This was a cross-sectional study of active duty service members who reported sustaining a TBI and provided self-report of neurological and psychological symptoms and provided blood.
Methods and procedures: An ultrasensitive single-molecule enzyme-linked immunosorbent assay was used to compare concentrations of Aβ in active duty service members with (TBI+; n = 53) and without (TBI–; n = 18) a history of TBI. Self-report and medical history were used to measure TBI occurrence and approximate the number of total TBIs and the severity of TBIs sustained during deployment.
Main outcomes and results: This study reports that TBI is associated with higher concentrations of Aβ40 (F1,68 = 6.948, p = 0.009) and a lower ratio of Aβ42/Aβ40 (F1,62 = 5.671, p = 0.020). These differences remained significant after controlling for co-morbid symptoms of post-traumatic stress disorder and depression.
Conclusions: These findings suggest that alterations in Aβ relate to TBIs and may contribute to chronic neurological symptoms.
Introduction
The Department of Defense estimates that over 300 000 active duty service members sustained one or more traumatic brain injuries (TBIs) during Operations Iraqi Freedom and Enduring Freedom, with many personnel experiencing multiple TBIs [Citation1]. TBIs significantly increase risk for the onset of neurodegenerative processes and chronic neurological symptoms, especially when sustained multiple times [Citation2] and increase the risk for neurological and psychological symptoms [Citation3]; however, this risk is not universal and is not well understood. Neurodegenerative conditions such as chronic traumatic encephalopathy (CTE) and Alzheimer’s disease (AD) are linked to TBIs [Citation4,Citation5], suggesting that TBIs involve neurological changes that may exacerbate some common neurodegenerative pathways that contribute to chronic neurological symptoms and deficits.
CTE is a tauopathy characterized by the deposition of hyperphosphorylated tau (p-tau) protein as neurofibrillary tangles, pathological processes that are also likely related to Aβ neuronal accumulations; however, this relationship is not definitive and is influenced greatly by age [Citation6]. Recently, deployment-related TBIs have been linked to high concentrations of total tau in the peripheral blood in a similar cohort of active duty service members with chronic symptoms, suggesting for the first time that these neurodegenerative processes can be detected in individuals with mild-to-moderate TBIs [Citation7]. This finding led the authors to question the role of Aβ in chronic symptoms following TBI in a sample of young active duty service members and to determine if use of a more sensitive detection method, which allows for Aβ measurement in peripheral blood, will provide evidence of Aβ involvement in TBI and chronic symptoms. In search of a minimally-invasive biomarker of TBI, there is interest in using peripheral blood concentrations of proteins to indicate injury severity. If a relationship between TBI and Aβ concentrations is found, it could possibly be used—along with tau—to create a prognostic biomarker for TBI patients.
Although PET studies continue to link tau accumulations to TBIs and chronic neurological symptoms [Citation8], the relationship between TBIs and formation of cerebral Aβ plaques has only been substantiated in subjects over the age of 50 years [Citation9]. In severe TBI, one study found decreased brain extracellular concentration of Aβ, a finding that may indicate TBI-induced suppression of neuronal activity [Citation10]. In peripheral blood samples, only one study has linked Aβ to TBIs, with higher concentrations in the blood during the acute recovery period after severe TBIs and the highest concentrations correlating with mortality risk [Citation11]. Aβ40 and Aβ42 are the two major isoforms of Aβ. They form from cleavage of the amyloid precursor protein and are the two amyloid peptides implicated in toxic plaque formation [Citation12,Citation13]. Aβ42 is considerably more hydrophobic and is, therefore, more likely to aggregate and form plaques than Aβ40 [Citation12]. There is interest in using the ratio of the two amyloid peptides as a biomarker of injury severity. Although the secondary injury pathway is not fully understood, there is evidence the amyloid peptide ratio could be an indicator of amyloid plaque formation or other TBI-related pathology. The plasma ratio of Aβ42/Aβ40 has been shown to correlate with AD pathology [Citation13–Citation16]. One study showed that an increase in Aβ40/42 ratios was associated with less plaque formation [Citation15]. Another study of patients with Alzheimer’s disease correlated cognitive decline with a decrease in Aβ42/40 ratios [Citation13].
Although these studies suggest a relationship between TBIs and Aβ peptides in the brain and periphery, no work has been undertaken to measure concentrations of Aβ in the peripheral blood of chronic TBI patients. This is due in part to the limits of previous technology in measuring Aβ peptides in the peripheral blood of patients with less severe TBIs. The concentrations of CNS proteins leaked to the plasma are in low concentrations that conventional analogue immunoassays are insufficiently sensitive enough to measure. The Simoa technology introduced by Quanterix Inc. (Lexington, MA) uses femtoliter-sized wells capable of trapping individual molecules and using a digital readout of beads to determine if they are bound to the target molecule—Aβ in this case. This ultrasensitive technology allows for detection of Aβ with increased sensitivity—over 1000 times more sensitive than conventional assays.
This exploratory study aims to fill a gap in the literature about chronic Aβ40 and Aβ42 concentrations in TBI patients by utilizing the novel Simoa methodology. It is hypothesized there will be a significant difference in concentrations of Aβ40 and Aβ42 between those who had a TBI (TBI+) and those who did not have a TBI (TBI–). If these differences are found, it may play a role in establishing Aβ as a peripheral biomarker that can help track central neurodegenerative processes related to chronic neurological symptoms following TBI. This line of research is necessary to determine the role of Aβ40 and Aβ42 in chronic neurological symptoms in a younger cohort who have often sustained multiple TBIs during deployment.
Methods
Participants
This was a prospective observational assessment of 71 active duty personnel who volunteered and consented to be in a larger research study at Madigan Army Medical Center that examined symptoms and biomarkers related to deployment. In the total study, there were a 172 participants who provided clinical information. For this sub-study, participants who provided blood and matched TBI cases to controls in demographics and clinical characteristics were included. This study was approved by the Institutional Review Board at Madigan Army Medical Center in Tacoma, WA. The patients provided voluntary informed consent and completed self-report questionnaires. The Warrior Administered Retrospective Casualty Assessment Tool (WARCAT) was used to determine if participants in this sample had a TBI, as well as the number of TBIs and the severity of the TBI. Controls were subjects who were similar to cases but did not report a TBI determined by the WARCAT. All participants had deployed within 16 months prior to sample collection. Exclusion criteria included the following: (1) history of drug or alcohol abuse in the previous year, (2) current severe medical conditions requiring chronic treatment (e.g. cancer, diabetes, HIV, autoimmune disorders) or a severe psychiatric condition (i.e. schizophrenia, bipolar disorder) and (3) severe neurological disorders (e.g. multiple sclerosis, seizure disorders, history of stroke). This study was approved by the Institutional Review Board (#145947) and informed consent was obtained from each individual prior to any baseline measurements.
Procedures for determining TBI groups
Fifty-three individuals with a history of TBI (TBI+) were identified by either self-reporting a TBI on the Warrior Administered Retrospective Casualty Assessment Tool (WARCAT) or by having a documented TBI in their medical record. Eighteen participants without a medical history of TBI in their records or a self-reported TBI on the WARCAT were classified as controls (TBI–). The military medical record was used to extract diagnosis of TBI and/or treatment for TBI during deployment. The WARCAT assesses war-related and post-deployment injuries by investigating the mechanism of injury as well as by asking about immediate symptoms or loss of consciousness after injury, indications that a subject sustained a TBI. Controls were matched to TBI cases as much as possible on critical variables that would have influenced biomarkers, including the following: age, gender, ethnicity, time since deployment and military rank.
Collection methods
Plasma samples were obtained from non-fasting subjects into EDTA tubes, transported on ice, centrifuged, aliquoted and frozen at –80°C. Subject availability varied such that collection times ranged from 9:00 a.m. to 4:00 p.m. (mean = 11:36 a.m.; SD = 1 hour, 54 minutes). Samples were thawed and analysed in a single batch.
Measures
Aβ40 and Aβ42 concentrations in plasma samples were analysed using Simoa, a high-definition-1 analyser, which is a paramagnetic bead-based ELISA. The kit includes a monoclonal anti-Aβ40 and 42 capture antibody directed to the N-terminus (Covance 6E10) and a biotinylated detector with a monoclonal anti-Aβ40 and Aβ42 antibody (ADx Neurosciences ADx and Invitrogen H31L21, respectively) directed to the C-terminus (Quanterix, Inc.) [Citation11]. The Aβ40 and Aβ42 assays have a low limit of detection of 0.76 pg ml–1 and 0.034 pg ml–1, respectively. The reported CV (intra- and inter-plate) values were below 10% for both Aβ40 and Aβ42. Samples with a CV value higher than 10% were excluded from analysis.
Symptoms of PCS were measured using the Neurobehavioural Symptom Inventory (NSI), a self-report measure evaluating 22 symptoms (e.g. balance, nausea and headache). The presence and severity of each symptom is evaluated on a 5-point scale: none, mild, moderate, severe, very severe). A total score is obtained by summation of each symptom score (range = 0–88), where a higher total score indicates higher symptom severity. The NSI has high internal consistency (total alpha = 0.95; sub-scale alpha = 0.88–0.92) and reliability (r = 0.88–0.93) [Citation17].
PTSD symptoms were measured using the PTSD Checklist Military Version (PCL-M), which has a score ranging from 0–80. Higher scores indicate higher severity, and a score over 50 indicates a clinical diagnosis of PTSD [Citation18].
Depression symptoms were measured using the Quick Inventory of Depressive Symptomatology (QIDS), which has a score ranging from 0–27. Higher scores indicate high severity and a score over 13 indicates a clinical diagnosis of depression [Citation19].
Statistical analysis
Descriptive statistics for all demographic and clinical variables were calculated using SPSS Statistics (IBM SPSS Inc., Chicago, IL; ). Comparisons were made between the two groups using Chi-square for categorical variables, analysis of variance (ANOVA) for continuous variables and analysis of covariance (ANCOVA) when adjusting for covariates. ANOVA models were used to compare Aβ40 and Aβ42 concentrations and the ratio of Aβ42 and Aβ40 in active duty service members who sustained a TBI to controls with no TBI. Lastly, the Spearman rank correlation coefficient was used to determine correlations between Aβ40 and Aβ42 concentrations, the Aβ42/Aβ40 ratio, injury characteristics, and clinical symptoms of PTSD and depression.
Table I. Demographics and clinical characteristics for TBI– and TBI+ groups (n = 71).
Results
This sample consisted of 71 active duty service members who were primarily male and who had deployed within the previous 16 months to combat stations (). The mean time since latest deployment was 9.7 months (SD = 5.0) and the mean time since the most severe deployment-related TBI was 20.6 months (SD = 12.3). The TBI+ (n = 53) and TBI– (n = 18) groups did not differ significantly in age or gender. The TBI+ group had significantly more symptoms of PTSD, depression and PCS. In the TBI+ group, the mean number of TBIs was 3.89 (SD = 1.93) and ~ 56.6% had a loss of consciousness ranging from less than a minute to 20 minutes, with 9.4% having a loss consciousness of more than 20 minutes in duration (). Approximately 38.43% of the TBI+ group had a medical diagnosis of TBI or had documentation of receiving care for a TBI during deployment. These ICD codes included the following: 85.40 or 959.01.
Table II. TBI characteristics according to the WARCAT.
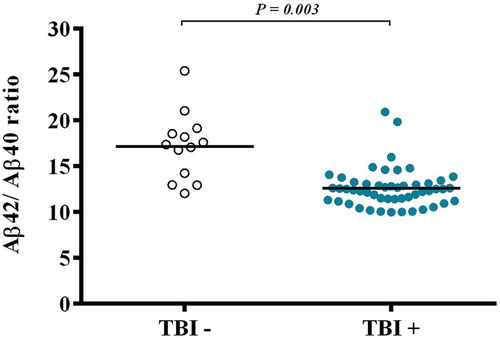
Concentrations of Aβ40 were significantly higher in the TBI+ group than the TBI– group (F1,68 = 6.948, p = 0.009; (a)). Concentrations of Aβ42 tended to be higher in the TBI+ group than the TBI– group (F1,64 = 2.979, p = 0.089; (b)) in active duty service members The ratio of Aβ42/Aβ40 was also significantly different between the groups (F1,62 = 5.671, p = 0.020), with the ratio being significantly lower in the TBI+ group (). In an ANCOVA model controlling for PTSD and depression, Aβ40 (F1,68 = 3.32, p = 0.025) and the ratio of Aβ42/Aβ40 (F1,62 = 5.01, p = 0.03) remained significantly different between the TBI+ and TBI– groups. Symptomatology of PTSD and depression was not related to concentrations of Aβ40 or Aβ42 in a correlation analysis of the ratio of Aβ42/Aβ40.
Figure 1. (a) Concentrations of Aβ40 were significantly different (F1,68 = 6.948, p = 0.009), with higher concentrations in the TBI+ group compared to TBI– controls. (b) Concentrations of Aβ42 tended to be different (F1,64 = 2.979, p = 0.089), with a tendency to have higher concentrations in the TBI+ group compared to TBI– controls.
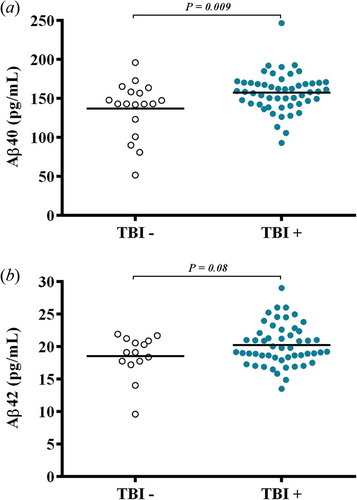
Figure 2. The ratio of Aβ42/Aβ40 was also significantly different between the groups (F1,62 = 5.671, p = 0.020), with the ratio being significantly lower in the TBI+ group compared to TBI– controls.
Discussion
This study reports for the first time that Aβ40 is elevated and the Aβ42/Aβ40 ratio is lower in the blood of young males with a history of deployment-related TBIs compared to those without a TBI. This finding provides insight into the mechanisms related to the onset of chronic neurological symptoms, which may lead to neurodegenerative processes later in life and the onset of CTE- and AD-related deficits [Citation3,Citation20]. Neurodegenerative processes, such as AD and CTE, include senile plaques and intracellular neurofibrillary tangles formed by hyper-phosphorylated tau in older individuals with Aβ accumulation [Citation9]. Previously, it has been shown that, in active duty service members, tau is also chronically elevated in individuals who have sustained a TBI compared to controls [Citation7]. The current study adds a novel investigation concerning peripheral Aβ concentrations in chronic TBI patients from a similar military cohort. This report now shows that Aβ also remains chronically elevated for months to years after sustaining a TBI, suggesting that the neuropathology may be related to that of AD and CTE.
Although one limitation to this study is that it is reliant on peripheral samples that cannot be generalized to central samples and processes, other studies suggest that these peripheral elevations result from TBI-induced axonal injuries [Citation21,Citation22]. Elevated Aβ concentrations suggest that there is an on-going neural injury, potentially leading to neurodegeneration, and that blood–brain barrier disruption may persist for long periods following TBI. While it is highly controversial whether amyloid plaques are toxic, there is ample experimental evidence to suggest that oligomeric forms of Aβ may be synaptotoxic and neurotoxic, promoting disruptions in cellular membrane and mitochondrial function and activating microglia to induce neuroinflammatory processes, with Aβ42 being more toxic than Aβ40 [Citation23–Citation30]. The plasma ratio of Aβ42/Aβ40 has been shown to be consistently associated with AD pathology [Citation13–Citation16] and plaque formation [Citation15]. Moreover, a study with patients with Alzheimer’s disease correlated cognitive decline with a decrease in Aβ42/40 ratios [Citation13]. Similarly, in this study, the ratio of Aβ42/Aβ40 was significantly lower in the TBI+ group. Although this finding suggests the ratio of Aβ42/40 could be a useful biomarker of neurodegeneration, there are limitations to the Simoa technology used in this study. It is possible that, because Aβ42 is more hydrophobic and prone to aggregation, Simoa’s ability to differentiate between oligomers and monomers is impeded, thereby resulting in an under-estimation of the peripheral concentrations of Aβ42 and affecting the ratio [Citation31]. This will require further study to differentiate the meaning of the ratio in chronic TBI and replication of this study using digital immunoassay technology advances.
The findings are also consistent with previous findings in TBI and CTE research [Citation32–Citation36]. A recent study using amyloid PET scans showed that patients who sustained a mild TBI, an average of 6 years prior to the study, had increased concentrations of amyloid, and these accumulations were associated with cognitive impairments in a sample of older subjects [Citation37]. In this sample of younger subjects, it was seen that individuals with TBIs, irrelevant of symptoms, have a higher concentration of Aβ40. In studies of CTE, it is reported that, in the earlier stages, the predominant symptoms are neurobehavioural and then progress to cognitive deficits [Citation38] and that cognitive symptoms only occur in older individuals with CTE, suggesting that age may be a contributor to Aβ accumulation and cognitive symptoms [Citation37]. Interestingly, epidemiological studies suggest that a TBI stimulates the accumulation of central Aβ and considerably shortens the time of onset of AD from 18 to 10 years [Citation20]. Indeed, a single TBI may result in neurofibrillary tangle and senile plaque formation that can promote the development of neurodegenerative processes and neurological disorders [Citation39]. Taken together with previous studies, the findings suggest that active duty service members exposed to TBI during combat may be at higher risk for developing AD or CTE. This diagnosis could present a window of opportunity for studying mechanisms of the neurodegenerative process and help slow the progression. Thus, in this younger cohort, it is likely that the Aβ-related neurodegenerative processes have not yet resulted in substantial cognitive-related deficits and it may be that these peripheral elevations are early indicators of neurodegeneration. This study suggests that these younger cohorts are followed over time to determine the relationship between AB and cognitive symptoms.
Although these findings suggest future implications, some limitations to this study are the sample size and lack of follow-up data. Additional studies with larger sample sizes and more long-term follow-up are necessary to determine the role of Aβ in TBI and chronic symptoms. In addition, the method to measure Aβ in a peripheral sample provides an opportunity to examine possible links in patients with non-severe traumatic brain injuries; however, these findings do not provide the necessary information to understand the role of neuronal Aβ accumulation in the formation of neurofibrillary tangles and risk for CTE without corroborating evidence in the form of imaging results or cerebrospinal fluid. Another limitation is the variable time of sample collection following head injury. Additional efforts should be made for prospective studies that acquire samples immediately after TBI and follow them longitudinally. Long-term prospective studies would help elucidate the temporal changes in post-TBI amyloid accumulations, which in turn would provide a clearer understanding of injury-catalysed biochemical cascades and support the use of these proteomic markers as prognostic tools.
Declaration of interest
This study was funded, in part, by the Center for Neuroscience and Regenerative Medicine (Grant 60855). This study was funded by the intramural research program of the National Institutes of Nursing Research. The authors report no conflicts of interest. The opinions and assertions in this manuscript are those of the authors and do not necessarily represent those of the Department of the Army, Department of Defense, US Government, or the Center for Neuroscience and Regenerative Medicine.
References
- Department of Defense worldwide TBI numbers [Internet]. D.a.V.B.I. Center; 2014. Available from http://dvbic.dcoe.mil/dod-worldwide-numbers-tbi.
- Reid MW, Miller KJ, Lange RT, Cooper DB, Tate DF, Bailie J, Brickell TA, French LM, Asmussen S, Kennedy JE. A multisite study of the relationships between blast exposures and symptom reporting in a post-deployment active duty military population with mild traumatic brain injury. Journal of Neurotrauma 2014;31:1899–1906.
- Yurgil KA, Barkauskas DA, Vasterling JJ, Nievergelt CM, Larson GE, Schork NJ, Litz BT, Nash WP, Baker DG, Marine Resiliency Study. Association between traumatic brain injury and risk of posttraumatic stress disorder in active-duty Marines. JAMA Psychiatry 2014;71:149–157.
- Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, Decarli C, et al. Head injury and the risk of AD in the MIRAGE study. Neurology 2000;54:1316–1323.
- BL Plassman, RJ Havlik, DC Steffens, MJ Helms, TN Newman, Drosdick D, Phillips C, BA Gau, KA Welsh-Bohmer, JR Burke, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology 2000;55:1158–1166.
- Mckee AC, Stein TD, Kiernan PT, Alvarez VE. The neuropathology of chronic traumatic encephalopathy. Brain Pathology 2015;25:350–364.
- Olivera A, Lejbman N, Jeromin A, French LM, Kim HS, Cashion A, Mysliwiec V, Diaz-Arrastia R, Gill J. Peripheral total tau in military personnel who sustain traumatic brain injuries during deployment. JAMA Neurology 2015;72:1109–1116.
- JR Barrio, GW Small, Wong KP, SC Huang, Liu J, DA Merrill, CC Giza, RP Fitzsimmons, Omalu B, Bailes J, et al. In vivo characterization of chronic traumatic encephalopathy using [F-18]FDDNP PET brain imaging. Proceedings of the National Academy of Sciences (USA) 2015;112:E2039–E2047.
- Stein TD, Montenigro PH, Alvarez VE, Xia W, Crary JF, Tripodis Y, Daneshvar DH, Mez J, Solomon T, Meng G. Beta-amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathologica 2015;130:21–34.
- Magnoni S, Brody DL. New perspectives on amyloid-beta dynamics after acute brain injury: moving between experimental approaches and studies in the human brain. Archives of Neurology 2010;67:1068–1073.
- Mondello S, Buki A, Barzo P, Randall J, Provuncher G, Hanlon D, Wilson D, Kobeissy F, Jeromin A. CSF and plasma amyloid-beta temporal profiles and relationships with neurological status and mortality after severe traumatic brain injury. Science Reports 2014;4:6446.
- Sun X, Chen WD, Wang YD. Beta-amyloid: the key peptide in the pathogenesis of Alzheimer’s disease. Frontiers in Pharmacology 2015;6:221.
- Seppala TT, Herukka SK, Hanninen T, Tervo S, Hallikainen M, Soininen H, Pirttila T. Plasma Abeta42 and Abeta40 as markers of cognitive change in follow-up: a prospective, longitudinal, population-based cohort study. Journal of Neurology, Neurosurgery and Psychiatry 2010;81:1123–1127.
- Dumurgier J, Schraen S, Gabelle A, Vercruysse O, Bombois S, Laplanche JL, Peoc’h K, Sablonniere B, Kastanenka KV, Delaby C, et al. Cerebrospinal fluid amyloid-beta 42/40 ratio in clinical setting of memory centers: a multicentric study. Alzheimers Research & Therapy 2015;7:30.
- Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. Journal of Neuroscience 2005;25:2803–2810.
- Jan A, Gokce O, Luthi-Carter R, Lashuel HA. The ratio of monomeric to aggregated forms of Abeta40 and Abeta42 is an important determinant of amyloid-beta aggregation, fibrillogenesis, and toxicity. Journal of Biological Chemistry 2008;283:28176–28189.
- King PR, Donnelly KT, Donnelly JP, Dunnam M, Warner G, Kittleson CJ, Bradshaw CB, Alt M, Meier ST. Psychometric study of the Neurobehavioral Symptom Inventory. Journal of Rehabilitation Research and Development 2012;49:879–888.
- Wilkins KC, Lang AJ, Norman SB. Synthesis of the psychometric properties of the PTSD checklist (PCL) military, civilian, and specific versions. Depression and Anxiety 2011;28:596–606.
- Trivedi MH, Rush AJ, Ibrahim HM, Carmody TJ, Biggs MM, Suppes T, Crismon ML, Shores-Wilson K, Toprac MG, Dennehy EB, et al. The Inventory of Depressive Symptomatology, Clinician Rating (IDS-C) and Self-Report (IDS-SR), and the Quick Inventory of Depressive Symptomatology, Clinician Rating (QIDS-C) and Self-Report (QIDS-SR) in public sector patients with mood disorders: a psychometric evaluation. Psychological Medicine 2004;34:73–82.
- Nemetz PN, Leibson C, Naessens JM, Beard M, Kokmen E, Annegers JF, Kurland LT. Traumatic brain injury and time to onset of Alzheimer’s disease: a population-based study. American Journal of Epidemiology 1999;149:32–40.
- MacDonald CL, Johnson AM, Cooper D, Nelson EC, Werner NJ, Shimony JS, Snyder AZ, Raichle ME, Witherow JR, Fang R, et al. Detection of blast-related traumatic brain injury in U.S. mlitary personnel. New England Journal of Medicine 2011;364:2091–2100.
- Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, Mclean AJ. Staining of amyloid precursor protein to study axonal damage in mild head injury. Lancet 1994;344:1055–1056.
- Kumar A, Singh A, Ekavalit. A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacology Reports 2015;67:195–203.
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002;416:535–539.
- Berman DE, Dall’armi C, Voronov SV, Mcintire LB, Zhang H, Moore AZ, Staniszewski A, Arancio O, Kim TW, Di Paolo G. Oligomeric amyloid-beta peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nature Neuroscience 2008;11:547–554.
- Claeysen S, Cochet M, Donneger R, Dumuis A, Bockaert J, Giannoni P. Alzheimer culprits: cellular crossroads and interplay. Cellular Signaling 2012;24:1831–1840.
- Koffie RM, Hashimoto T, Tai HC, Kay KR, Serrano-Pozo A, Joyner D, Hou S, Kopeikina KJ, Frosch MP, Lee VM, et al. Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-beta. Brain 2012;135:2155–2168.
- Parihar MS, Brewer GJ. Amyloid-beta as a modulator of synaptic plasticity. Journal of Alzheimers Disease 2010;22:741–763.
- Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M. Association of microglia with amyloid plaques in brains of APP23 transgenic mice. American Journal of Pathology 1999;154:1673–1684.
- Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochemistry Society Transactions 2002;30:552–557.
- Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ, Holtzman DM. Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science 2008;321:1221–1224.
- Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neuroscience Letters 1993;160:139–144.
- Jordan BD, Kanik AB, Horwich MS, Sweeney D, Relkin NR, Petito CK, Gandy S. Apolipoprotein E epsilon 4 and fatal cerebral amyloid angiopathy associated with dementia pugilistica. Annals of Neurology 1995;38:698–699.
- Roberts GW, Gentleman SM, Lynch A, Graham DI. beta A4 amyloid protein deposition in brain after head trauma. Lancet 1991;338:1422–1423.
- Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI. Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. Journal of Neurology, Neurosurgery & Psychiatry 1994;57:419–425.
- Teasdale GM, Nicoll JA, Murray G, Fiddes M. Association of apolipoprotein E polymorphism with outcome after head injury. Lancet 1997;350:1069–1071.
- Yang ST, Hsiao IT, Hsieh CJ, Chiang YH, Yen TC, Chiu WT, Lin KJ, Hu CJ. Accumulation of amyloid in cognitive impairment after mild traumatic brain injury. Journal of Neurological Sciences 2015;349:99–104.
- Stern RA, Daneshvar DH, Baugh CM, Seichepine DR, Montenigro PH, Riley DO, Fritts NG, Stamm JM, Robbins CA, Mchale L, et al. Clinical presentation of chronic traumatic encephalopathy. Neurology 2013;81:1122–1129.
- Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathology 2012;22:142–149.