Abstract
Analytical methods developed to sample and characterize ambient organic aerosols often face the trade-off between long sampling times and the loss of detailed information regarding specific chemical species present. In the work presented here, high-time resolution ambient measurements (3.5 minutes) are achieved by using the Photoionization Aerosol Mass Spectrometer (PIAMS) in conjunction with the mini-Versatile Aerosol Concentration Enrichment System (m-VACES). Aerosol sampling took place for one week at State of Delaware Air Quality Monitoring Site in Wilmington, Delaware in June 2006. The soft, universal ionization scheme of PIAMS allows for identification of various chemical compounds by a signature ion, often the molecular ion. Meteorological data was used to link some species, such as palmitic and oleic acid, to potential sources; whereas other compounds, like PAHs, have no significant wind dependence. Some classes, including n-alkanes and n-alkanoic acids, are shown to be in abundance during late night/early morning hours, potentially due to the combined effects of temperature inversion, and fresh emissions in a cooler environment. The high-time resolution data shows that rapid concentration changes of a common individual species can be lost with traditional bulk sampling, and a time resolution of 30 minutes is suggested to accurately represent these changes.
INTRODUCTION
There is a significant concern over the impact of ambient aerosols. Particulate matter (PM) in the atmosphere is known to affect visibility (CitationSisler and Malm 1994) and influence global climate (CitationCharlson et al. 1992; CitationMaria et al. 2004). Human exposure can cause a variety of detrimental health effects (CitationMills et al. 2005; CitationSamet et al. 2000; CitationZanobetti and Schwartz 2006). Much of ambient aerosol, whether biogenic or anthropogenic, is comprised of organic compounds. Depending on the location, organic matter can comprise anywhere from 10–70% of the total particle mass in the air (CitationTurpin et al. 2000). To better understand the organic fraction, numerous studies have used filter-based sampling with gas-chromatography mass-spectrometry (GC/MS) to identify and quantify emissions from specific sources (e.g., CitationRogge et al. 1991; CitationSchauer et al. 1999a) and perform source apportionment (e.g., CitationRobinson et al. 2006b; CitationSchauer et al. 1996). Despite these advances, much remains unknown. The large number of potential compounds can overwhelm a chromatographic separation, causing unresolved peaks. The low concentration levels of individual compounds (usually less than ∼ 10 ng/m3) require long sampling times, often 3–24 hours, to exceed detection limits. With this low-time resolution data, information is lost regarding short-term changes in local aerosol concentration.
Variations on the traditional GC-MS method are designed to better the time-resolution and cut down on sample preparation. A time-resolution of 1 hour has been achieved with a thermal desorption GC/MS-FID, or TAG (CitationWilliams et al. 2006). TAG is an automated technique that can collect compound specific data similar to filter-based GC-MS measurements. When combined with meteorological data, the faster time-resolution measurements are more suitable to describe local primary and secondary organic sources (CitationWilliams et al. 2007).
Alternative techniques, notably aerosol mass spectrometers, also have been developed to address the shortcomings of traditional sampling methods (CitationHinz and Spengler 2007; CitationMurphy 2007; CitationNash et al. 2006). These instruments can achieve time resolution on the order of a few minutes or less. The Aerodyne Research, Inc. Aerosol Mass Spectrometer (AMS) is one of the more common real-time instruments in use (CitationCanagaratna et al. 2007; CitationDeCarlo et al. 2006). The AMS is based on 70 eV electron ionization (EI), which yields reproducible fragmentation patterns, allowing the development of algorithms to resolve general organic aerosol classes, namely hydrocarbon-like (HOA) and oxygenated organic aerosol (OOA)(CitationZhang et al. 2005a, Citation2005b). Detailed information can be extracted for some stable compounds, such as polycyclic aromatic hydrocarbons (PAHs)(CitationMarr et al. 2006). However, most organic molecules fragment extensively with 70 eV EI, and information regarding individual species is lost.
Another approach has been to use a “soft” ionization method to limit fragmentation, which in turn aids identification and measurement of individual species. Instruments utilizing two-step laser desorption/laser ionization, in conjunction with a rotating drum-impactor can achieve a time-resolution of 20 minutes between samples (CitationEmmenegger et al. 2005). The resonance enhanced multiphoton ionization technique, while highly sensitive to aromatic compounds, cannot be used for aliphatic compounds (CitationHaefliger et al. 2000). Other techniques such as photoelectron resonance capture ionization (CitationLaFranchi and Petrucci 2004), filter-based infrared laser desorption—single photon ionization mass spectrometry (CitationFerge et al. 2005) and hybrid-thermal desorption/photoionization mass spectrometry (CitationMitschke et al. 2005; CitationStreibel et al. 2006), aerosol chemical ionization mass spectrometry (CitationHearn and Smith 2004), and the photoionization aerosol mass spectrometer (PIAMS)(CitationOktem et al. 2004) use soft ionization methods that can be applied to both aliphatic and aromatic species. These instruments have the potential for highly time-resolved measurements, but they cannot distinguish isobaric compounds.
The method used in this study, PIAMS, achieves soft ionization by single-photon ionization (SPI). The photon energy of 10.5 eV (118 nm) is just above the ionization energy of most organic molecules. Additionally, the instrument is sensitive, transportable, and automated for field measurements. Particles are delivered directly to the source region, so no sample work-up is required. Previously, PIAMS has been used to sample common organic emissions (CitationOktem et al. 2004), characterize soot particles in a flame (CitationOktem et al. 2005), monitor the rate of cholesterol ozonolysis (CitationDreyfus et al. 2005), and characterize SOA formation from monoterpene ozonolysis (CitationHeaton et al. 2007; CitationTolocka et al. 2006).
Here, PIAMS was used to sample ambient organic aerosol at the State of Delaware Air Quality Monitoring Site in Wilmington, Delaware. When used with the ca. 20× aerosol enrichment provided by the mini-Versatile Aerosol Concentration Enrichment System (m-VACES)(CitationGeller et al. 2005; CitationNing et al. 2006), PIAMS can characterize individual compounds in ambient aerosol down to about 1 ng/m3 with a time resolution of less than 4 minutes. The results from these initial field measurements provide information on specific organic species and how they change as a function of time and wind direction, giving further insight to the complex behavior of organic aerosols in urban air.
EXPERIMENTAL
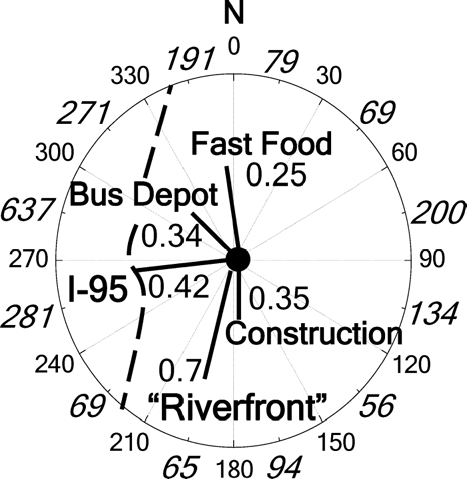
Aerosol sampling was performed at the State of Delaware Air Quality Monitoring Site on Martin Luther King Boulevard in Wilmington, Delaware from Sunday, June 4 to Sunday, June 11, 2006. The site is located in a mostly urban area, within a short distance to a major roadway (Interstate-95), and the Riverfront, an entertainment hub consisting of shops, restaurants, and a minor-league baseball stadium. Relative locations to the sampling site are shown in . All instrumentation was housed in a trailer at ∼ 20°C. The outside temperature ranged from 14–26°C with very little precipitation. Wind speed and direction were measured every minute on the premises. All wind directions were sampled over the course of the study; however wind from the northwest was the most predominant, especially later in the week.
FIG. 1 Relative location (km) of local organic aerosol emission sources to the sampling site. Several building construction sites were located to the south and southeast. The numbers in italics around the inside of the circle represent the number of samples from each wind direction during the study.
Ambient aerosol was pulled through a PM 10 inlet at 30 lpm to the m-VACES (CitationGeller et al. 2005) (). The concentrated aerosol stream exits at a rate of 1.5 lpm. The sample is then sent to PIAMS at 0.1 lpm; excess aerosol flow is sent to a condensation particle counter (CPC; TSI model 3025a, Shoreview, MN) to monitor aerosol enrichment from the m-VACES. PIAMS typically requires several picograms of material sampled through the inlet to obtain a robust signal. This amount can be obtained quickly with the current setup. For example, an ambient concentration of 1 ng/m3 for an individual organic species that is subsequently concentrated by a factor of 20 with m-VACES and sampled into the mass spectrometer at a rate of 0.1 lpm gives a sampling rate of 2 pg/min. The current system is more sensitive than that reported previously (CitationOktem et al. 2004), largely due to the use of a stainless steel probe for particle collection. To ensure an adequate signal level for a variety of species, the sample collection time was set to 2 minutes, immediately followed by sample analysis (20 seconds, 200 recorded mass spectra) and probe cleaning (70 seconds) to remove any artifacts. A full sampling cycle was completed every 3.5 minutes and the instrument ran nearly continuously for 24-hour spans. Typically, sampling would be suspended for ∼1 hour each day for general maintenance. This allowed for at least 320 samples analyzed per day.
FIG. 2 Schematic of experimental setup.
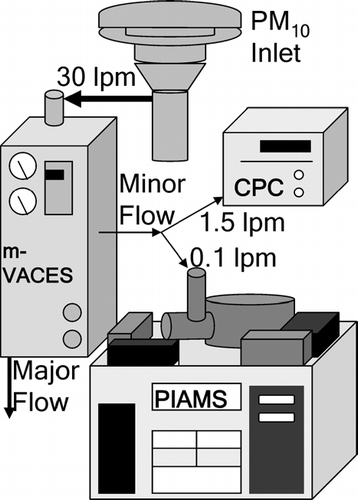
PIAMS has been described in detail previously (CitationOktem et al. 2004). Briefly, the aerosol enters through a computer-controlled valve and into an aerodynamic lens assembly, which focuses the sample into a particle beam (CitationLiu et al. 1995a, b). The resulting beam is deposited onto a cooled, stainless steel probe (∼ 0°C) located in the ion source region. When the collection period is complete, a 1064 nm Nd:YAG laser is pulsed at low energy (ca. 3 mJ total; 40 mJ/cm2) to irradiate the probe, rapidly heating its surface. The rapid temperature increase results in a flash vaporization of the deposited material. About 30 μ s after the desorption laser pulse, a second Nd:YAG laser fires to ionize the desorbed analyte. Ionization is performed by single photon ionization (SPI) with 10.5 eV (118 nm) radiation, obtained by frequency tripling the third harmonic of the Nd:YAG laser (CitationVan Bramer and Johnston 1990). The photoionization mass spectrum is obtained with a reflectron time-of-flight mass analyzer that allows the entire mass spectrum to be obtained for each laser pulse. The desorption/ionization sequence is repeated 200 times per sample at a 10 Hz repetition rate (20 seconds total) to allow all deposited material to be desorbed and ionized. The lasers are continuously fired for another 70 seconds for cleaning. The entire process is automated using LabVIEW software (National Instruments, Austin, TX).
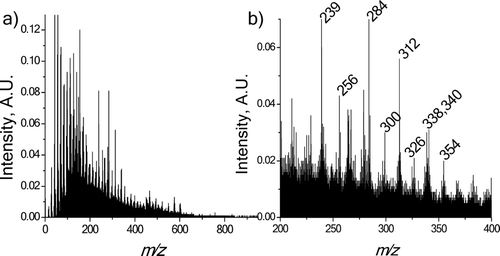
Over 2200 samples were analyzed in 180 available sampling hours. Data analysis was performed off-line after the measurement campaign was concluded. The 200 individual spectra taken after each 2-minute sampling period were averaged to give a single mass spectrum. Each averaged spectrum was then baseline corrected and binned to ± 0.5 Da around the central m/z. Finally, each binned spectrum was normalized by finding its unit vector. shows an example spectrum. Other spectra in the data set are similar in character, although the signal intensities of specific m/z ions change with time.
FIG. 3 (a) Example mass spectrum; results from 2 minute sample collection beginning at 8:58 PM on 6/8/06. (b) Same spectrum, zoomed in from 200–400 m/z. Majority of peaks in this region are due to molecular ions.
RESULTS AND DISCUSSION
Identification was based on molecular ion m/z, or in some cases the appearance of a signature fragment ion as reported in the literature. Nearly all of the suggested molecular ion identifications were based on species that are well-known, often abundant, primary organic emissions. Isobaric interference prevents definitive identifications. When the results for a particular m/z value are discussed, they should be understood to include both the expected dominant species and any potential isomers and isobars. While the ion signal intensities provide a quantitative measure of changes in concentration, determination of the absolute amount of each chemical component is not currently possible.
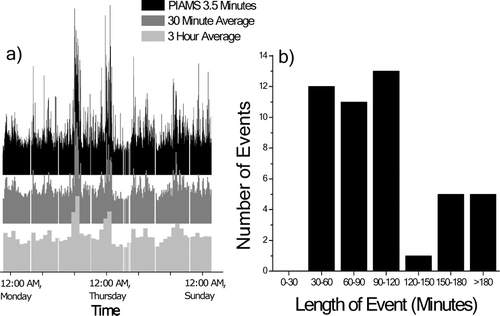
The highly time resolved PIAMS data set provides information concerning the concentration changes of individual organic compounds as a function of time or wind direction. A plot of signal vs. time with 3.5 minute time intervals shows significantly more detail than typical collection-based techniques. shows the time series for 256 m/z (corresponds to palmitic acid plus its isomers and isobars) along with averages over 30 minute and 3 hour intervals. Whereas 30 minute time-resolution can capture most of the significant concentration changes observed, 3 hour resolution loses all but general trends in the concentration. To illustrate this concept further, the duration of various high concentration events for 20 different m/z values (149, 191, 202, 240, 252, 256, 268, 270, 282, 284, 312, 326, 338, 352, 368, 382, 422, 438, 450, and 576) are shown in . These events were defined as any signal increase to ≥ 50% of the mean signal intensity. Concentration events occurring simultaneously for different m/z values were counted as one event. Most of these events were shorter than 180 minutes; with a large fraction lasting between 30 and 120 minutes. The distribution shown in suggests that a time resolution of 30 minutes will be sufficient to describe the behavior of ambient primary organic aerosols. Concentration spikes shorter than 30 minutes were seen throughout the dataset, but none more intense than the aforementioned threshold.
FIG. 4 a) Time series of palmitic acid (256 m/z) from 7:17 PM, June 8th to 7:09, June 10th, with different time resolutions. Slower time resolution, especially 3+ hours, loses much of the dynamic concentration changes seen in the PIAMS data. b) Number and length of high concentration particle events (signal peak ≥ 50% of mean intensity) for 20 different species.
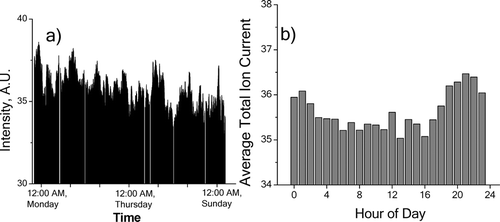
The total ion signal as a function of time, which represents changes in the total organic carbon aerosol loading, is shown in . The higher aerosol loading at night is likely the combination of several factors that cannot be independently distinguished at this time, including (1) decrease in mixing height, (2) decrease in temperature which favors partitioning of semivolatile species to the particle phase, and (3) high levels of human activity in the local area. A similar nighttime enhancement was seen by CitationPark et al. (2005) with hourly OC/EC measurements during the month of June 2002 at the Baltimore particulate matter supersite. Park et al. also observed a significant enhancement during the morning rush hour, which was not surprising as their measurement site was adjacent to a major Interstate highway. A morning rush hour enhancement is not observed in Wilmington, presumably because the measurement site is further from major traffic routes.
FIG. 5 a) Time series of the total ion signal during sampling period. Data points are smoothed by 8 point adjacent averaging to give a time resolution of approximately 30 min. b) Total ion signal averaged by time of day.
POINT SOURCES
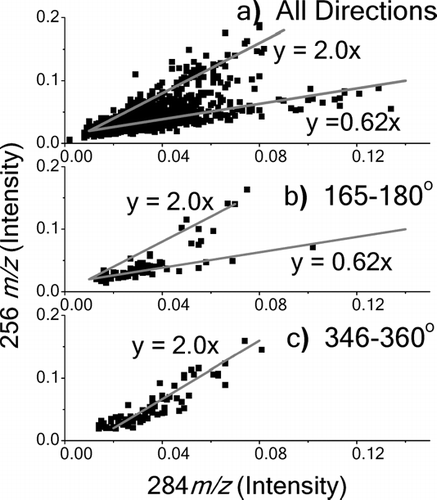
The most abundant compounds found during the sampling week were the n-alkanoic acids. These acids are emitted from nearly every major primary source of organic aerosol (CitationSchauer et al. 1996). Although the wind direction dependence of each individual species is unique, nearly all show a tendency for greater signal intensity from the southwest to southeast (). There is a significant diurnal dependence, with about 20% higher signal levels coming after 7 PM local time relative to earlier in the day (). The most prominent n-alkanoic acids, palmitic acid (C16H32O2, 256 m/z) and stearic acid (C18H36O2, 284 m/z), show different behavior. Although both are emitted from numerous sources, they are often linked with meat-cooking operations (CitationRobinson et al. 2006c; CitationRogge et al. 1991; CitationSchauer et al. 1999a). Few grill-based restaurants are located in the vicinity of the sampling site. A fast food restaurant is located a few blocks north/northwest, and to the south is the Riverfront, a shopping center with a few bar/grill establishments. Both of these compounds show a bias toward these directions in a wind-rose plot (). The average signal per hour reveals that both compounds peak in signal intensity in the early morning hours after midnight, and then decline through mid-day (). The magnitudes of the diurnal variations for these compounds are much greater than those for the other n-alkanoic acids. The signal intensities of palmitic and stearic acid for each sample are plotted against each other in a simple scatter plot in . Data points that combine to form a line indicate one source since both will rise and fall together (CitationRobinson et al. 2006b). In , two lines are drawn to indicate at least two separate sources. Furthermore, these plots were repeated using only data obtained from specific wind directions. and suggests one major source to the north, perhaps the fast food establishment, and possibly two sources to the south, one of which has a similar composition signature to that from the north.
FIG. 6 Wind and hourly time dependence of organic acids. Wind rose plot of average signal intensity vs. wind direction a) and diurnal dependence b) are shown for the averaged signal of several alkanoic acids (C15-30, without C16 & C18). Ions corresponding to palmitic and stearic acids (256 and 284 m/z, respectively) display much different wind c) and hourly d) trends. Signal from oleic acid (282 m/z) has similar wind e) and hourly f) dependences to palmitic and stearic acid. All six plots are averaged over the entire measurement period.
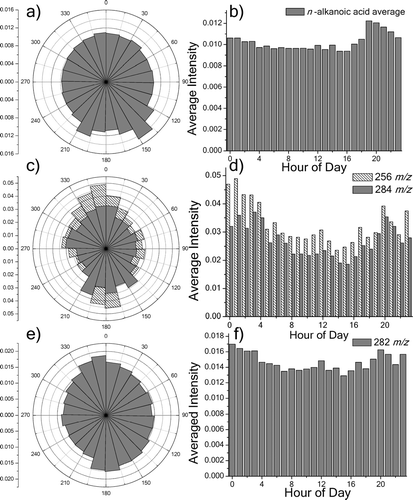
FIG. 7 Scatter plots of 256 m/z vs. 284 m/z. Lines are drawn as a guide to the eye. a) Plot of all measurements during week. b) Plot of measurements for a southerly wind direction (165–180°). c) Plot of measurements for a northerly wind direction (346–360°). Measurements for wind speeds less then 1 m/s are excluded from b) and c).
Two other components of meat-cooking aerosol were investigated in the dataset. Oleic acid (C18H34O2, 282 m/z) shows similar wind and time dependences to palmitic and stearic acids (, ). However, the north-south bias is less pronounced, suggesting that other sources contribute more to the concentration than palmitic and stearic acid. Cholesterol (C27H46O, 386 m/z) shows a different diurnal dependence, peaking in the daytime. However, no wind dependence is seen. This could be explained by the inherently low emission levels of cholesterol relative to the three acids above, along with the possibility that meat-less cooking gives the acids but not cholesterol. Although cholesterol has been used as a tracer in areas with a high-density of meat cooking operations (CitationRogge et al. 1991; CitationSchauer et al. 1996), cholesterol is also emitted from other sources, and not always has been linked to meat-cooking in source apportionment studies (CitationJaeckels et al. 2007).
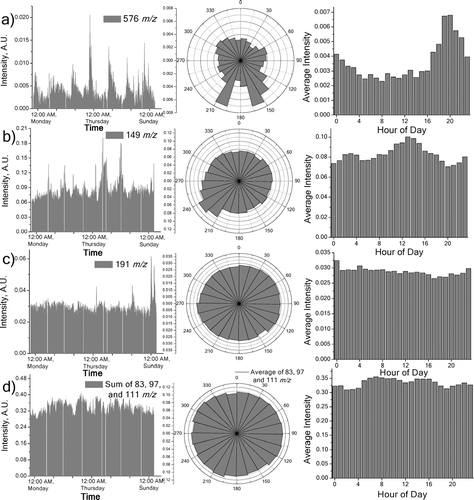
n-Alkanes are emitted from nearly ever major organic aerosol source, and sometimes they are the most abundant species (CitationRogge et al. 1997). Emissions from vegetative detritus (CitationRogge et al. 1993c) and plastic burning (CitationSimoneit et al. 2005) can show a dependence on odd or even numbered n-alkanes, suggesting its usefulness as a source tracer. The CPI (carbon preference index) from ions corresponding to C22-C37 n-alkanes is 0.93; thus no significant odd/even dependence is seen, nor is one seen in data isolated from wind directions pertaining to limited vegetation surrounding the site. Generally low wind-speed from the required directions, or simply the trivial amount of vegetation in the area would prevent leaf-abrasion aerosol from being produced at sufficient levels. Many m/z ratios corresponding n-alkanes, including 240, 310, 338, 366, 408, 422, 436, and 464 all show an increased proclivity to come from the east/southeast directions, and a smaller tendency from the southwest. It is of interest to report the appearance of signal at 576 m/z, here reported as hentetracontane, C41H84 (). n-Alkanes larger than C34 can be found in tire wear particles (CitationRogge et al. 1993b); however, hentetratcontane has not been reported in an ambient sample previously.
FIG. 8 Time series, wind, and hourly dependence data for various m/z values discussed in text. All time series are smoothed with 4-point data averaging. a) Hentetracontane (576 m/z) peaks during the nighttime hours, and has a bias towards the southwest and southeast. b) Phthalate ion 149 m/z shows daytime dependence, and proclivity to come from the southwest. c) Hopane fragment ion 191 m/z shows large peaks during the weekend nights. d) Sum of diesel exhaust related alkyl fragments 83, 97, and 111 m/z. A daytime enhancement is observed; the wind rose plot is the average of all three ions.
The similarities in data between the n-alkanes and n-alkanoic acids, both in wind and diurnal dependences, suggest their emission sources and behavior are similar. The diurnal dependence shown by both types of compounds support suggestions that emissions during the day are either mainly gas-phase or decay quickly from local oxidants (CitationRobinson et al. 2007; CitationStephanou 2005). The nighttime environment is more supportive of organic species in the aerosol phase, since the lower temperature allows primary organic emissions that undergo gas-particle partitioning to favor the particle phase.
Signal from 149, 167 and 279 m/z, a series of phthalate peaks, are seen in high levels of intensity throughout the week. Primary emissions of phthalates have been seen in high amounts during warm-temperature seasons in Sweden (CitationThuren and Larsson 1990) and suggested sources include plastics incineration (CitationSimoneit et al. 2004; CitationSimoneit et al. 2005; CitationStales et al. 1997). These ions show an enhanced intensity from the southwest, and a diurnal dependence that favors the daytime (). Possible sources from the southwest include a power plant, factory, recycling facility and traffic along Interstate-95. The strong afternoon dependence is potentially related to the higher temperature, resulting in a higher concentration of phthalates in the air. The increasing concentration during daylight hours could also indicate the resulting formation of secondary organic aerosol (SOA); phthalic acid has been identified as an SOA product (CitationLewandowski et al. 2007).
There is some concern of contamination from the limited amount of plastic sampling lines used from the PM inlet to the m-VACES and inside the m-VACES itself. However, the temperatures inside the trailer and the m-VACES were held constant, keeping sampling lines at a stable temperature. Furthermore, it is difficult to correlate the potential contamination with significant wind and time dependences shown in .
Levoglucosan (C6H10O5, 162 m/z) is the dominant particle phase emission of wood burning (CitationRogge et al. 1998; CitationSchauer et al. 2001; CitationSimoneit et al. 1999). As might be expected in the warmer summer weather, signal at 162 m/z (corresponding to levoglucosan) was not found in enhanced amounts relative to adjacent m/z values. This result was similar to that at the m/z values of the other common wood burning emissions, including abietic and dehydroabietic acid, syringaldehyde, vanillin, and isopimaric acid.
DISPERSE SOURCES
PAHs are carcinogenic compounds produced mainly from combustion sources such as vehicle exhaust, biomass burning and industrial processes (CitationHays et al. 2003; CitationLim et al. 2005; CitationMarr et al. 2006; CitationRobinson et al. 2006b; CitationRogge et al. 1993a). The sampling site is a short distance from a couple of major highways and is surrounded by various streets. Despite the presence of local traffic, no diurnal dependence on rush hour is evident. The high number of disperse sources around the site, combined with a semi-volatile nature of 2 and 3 ringed PAHs can explain the absence of any time or wind trends during the week. Even with relatively low signal intensities from the PAHs, a significant concentration spike was detected late in the week. This spike lasted 30 minutes, and the wind direction was the same before and after the event. This implies that it was caused by a short time period emission rather than a continuous plume that was temporarily swept to the site. It has been reported previously that PAH levels are often lower in the summer compared to other seasons (CitationEmmenegger et al. 2005; CitationLi et al. 2006).
Hopanes are a class of compounds strongly correlated to vehicle emissions (CitationRogge et al. 1993a; CitationSchauer et al. 1999b). They have a characteristic fragment ion at 191 m/z that is observed in the dataset. Much like the PAHs, no trend is seen regarding signal spikes at rush hour; however, significant spikes were seen in the early morning weekend hours (). Each of these high concentration episodes occurred when wind was coming in from the direction of Interstate-95. Traffic was not monitored during the course of the study, but it is possible that late night traffic events could have caused the signal increase. Another sampling session during cooler weather and/or maximum traffic period, like that seen during holiday travel, would provide further insight to the time trends seen here. Ambient hopane levels during the summer can be lower than other seasons (CitationRobinson et al. 2006a; CitationSubramanian et al. 2006).
Ions at 83, 97 and 111 m/z have previously been identified in PIAMS spectra as alkyl fragments from unburned fuel droplets in diesel exhaust (CitationOktem et al. 2004). These three ions correlate well to each other and have similar diurnal patterns (). Similar to the hopanes and PAHs discussed above, no wind dependence is seen in the data averaged over the sampling period. Unlike those compounds, the alkyl fragment ions are significantly enhanced in the daytime, especially before and after lunch hour. Furthermore, the alkyl fragment ions exhibit a different diurnal dependence from the n-alkane molecular ions. The time series of the alkyl fragment ions in shows a gradual increase during the week, then a decrease on the weekend. While the 24 hour wind rose plot in shows no directionality, a slight signal enhancement is observed in the direction of the Interstate and bus depot if only the daytime measurements are included.
CONCLUSIONS
Ambient organic aerosol measurements were achieved with a time resolution of 3.5 minutes using PIAMS in conjunction with the m-VACES. Measurements from an initial one week sampling campaign yielded detailed information concerning rapid concentration changes for a variety of pertinent organic compounds. This information was combined with meteorological data to correlate some chemical species with potential source locations. Some compounds, such as the n-alkanes and n-alkanoic acids, were observed at the highest levels during the night and showed significant wind direction dependence. Signal related to disperse sources such as car and diesel exhaust indicated little overall time and wind dependence. Based on the length of high-concentration particle events seen from a variety of different species, a minimum time resolution of 30 minutes is suggested to accurately describe most significant concentration changes. While absolute changes in ion signal intensities are directly related to absolute changes in ambient concentration, quantification of the actual amounts was not possible in the current study. In future work, quantification may be achieved when PIAMS measurements are referenced to other data such as GC-MS, OC/EC, and particle size distributions.
Acknowledgments
Phil Fine and Constantinos Sioutas are acknowledged for their help in providing and supporting work with the m-VACES. This work was funded by the United States Environmental Protection Agency (RD-83216601). Although the research described in this article was funded by the E.P.A., it has not been subjected to the Agency's required peer and policy review and therefore does not necessarily reflect the views of the Agency and no official endorsement should be inferred.
Related Research Data
REFERENCES
- Canagaratna , M. R. , Jayne , J. T. , Jimenez , J. L. , Allan , J. D. , Alfarra , M. R. , Zhang , Q. , Onasch , T. B. , Drewnick , F. , Coe , H. , Middlebrook , A. , Delia , A. , Williams , L. R. , Trimborn , A. M. , Northway , M. J. , DeCarlo , P. F. , Kolb , C. E. , Davidovits , P. and Worsnop , D. R. 2007 . Chemical and Microphysical Characterization of Ambient Aerosols with the Aerodyne Aerosol Mass Spectrometer . Mass Spectrom. Rev. , 26 : 185 – 222 .
- Charlson , R. J. , Schwartz , S. E. , Hales , J. M. , Cess , R. D. , Coakley , J. A. J. , Hansen , J. E. and Hofmann , D. J. 1992 . Climate Forcing by Anthropogenic Aerosols . Science , 255 : 423 – 430 .
- DeCarlo , P. F. , Kimmel , J. R. , Trimborn , A. , Northway , M. J. , Jayne , J. T. , Aiken , A. C. , Gonin , M. , Fuhrer , K. , Horvath , T. , Docherty , K. S. , Worsnop , D. R. and Jimenez , J. L. 2006 . Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer . Anal. Chem. , 78 : 8281 – 8289 .
- Dreyfus , M. A. , Tolocka , M. P. , Dodds , S. M. , Dykins , J. and Johnston , M. V. 2005 . Cholesterol Ozonolysis: Kinetics, Mechanism, and Oliogomer Products . J. Phys. Chem. A , 109 : 6242 – 6248 .
- Emmenegger , C. , Kalberer , M. , Samburova , V. and Zenobi , R. 2005 . High Time Resolution and Size-Segregated Analysis of Aerosol-Bound Polycyclic Aromatic Hydrocarbons . Environ. Sci. Technol. , 39 : 4213 – 4219 .
- Ferge , T. , Muhlberger , F. and Zimmermann , R. 2005 . Application of Infrared Laser Desorption Vacuum-UV Single-Photon Ionization Mass Spectrometry for Analysis of Organic Compounds from Particulate Matter Filter Samples . Anal. Chem. , 77 : 4528 – 4538 .
- Geller , M. D. , Biswas , S. , Fine , P. M. and Sioutas , C. 2005 . A New Compact Aerosol Concentrator for Use in Conjunction with Low Flow-Rate Continuous Aerosol Instrumentation . J. Aerosol Sci. , 36 : 1006 – 1022 .
- Haefliger , O. P. , Bucheli , T. D. and Zenobi , R. 2000 . Laser Mass Spectrometric Analysis of Organic Atmospheric Aerosols. 2. Elucidation of Urban Air Pollution Processes . Environ. Sci. Technol. , 34 : 2184 – 2189 .
- Hays , M. D. , Smith , N. D. , Kinsey , J. , Dong , Y. and Kariher , P. 2003 . Polycyclic Aromatic Hydrocarbon Size Distributions in Aerosols from Appliances of Residential Wood Combustion as Determined by Direct Thermal Desorption–Gc/Ms . J. Aerosol Sci. , 34 : 1061 – 1084 .
- Hearn , J. D. and Smith , G. D. 2004 . A Chemical Ionization Mass Spectrometry Method for the Online Analysis of Organic Aerosols . Anal. Chem. , 76 : 2820 – 2826 .
- Heaton , K. J. , Dreyfus , M. A. , Wang , S. and Johnston , M. V. 2007 . Oligomers in the Early Stage of Biogenic Secondary Organic Aerosol Formation and Growth . Environ. Sci. Technol. , 41 : 6129 – 6136 .
- Hinz , K.-P. and Spengler , B. 2007 . Instrumentation, Data Evaluation and Quantification in On-Line Aerosol Mass Spectrometry . J. Mass Spectrom. , 42 : 843 – 860 .
- Jaeckels , J. M. , Bae , M. S. and Schauer , J. J. 2007 . Positive Matrix Factorization (Pmf) Analysis of Molecular Marker Measurements to Quantify the Sources of Organic Aerosols . Environ. Sci. Technol. , 41 : 5763 – 5769 .
- LaFranchi , B. W. and Petrucci , G. A. 2004 . Photoelectron Resonance Capture Ionization (Perci): A Novel Technique for the Soft-Ionization of Organic Compounds . J. Am. Soc. Mass Spectrom. , 15 : 424 – 430 .
- Lewandowski , M. , Jaoui , M. , Kleindienst , T. E. , Offenberg , J. H. and Edney , E. O. 2007 . Composition of Pm2.5 During the Summer of 2003 in Research Triangle Park, North Carolina . Atmos. Environ. , 41 : 4073 – 4083 .
- Li , M. , McDow , S. R. , Tollerud , D. J. and Mazurek , M. A. 2006 . Seasonal Abundance of Organic Molecular Markers in Urban Particulate Matter from Philadelphia, PA . Atmos. Environ. , 40 : 2260 – 2273 .
- Lim , M. C. H. , Ayoko , G. A. , Morawska , L. , Ristovski , Z. D. and Rohan Jayaratne , E. 2005 . Effect of Fuel Composition and Engine Operating Conditions on Polycyclic Aromatic Hydrocarbon Emissions from a Fleet of Heavy-Duty Diesel Buses . Atmos. Environ. , 39 : 7836 – 7848 .
- Liu , P. , Ziemann , P. J. , Kittelson , D. B. and McMurry , P. H. 1995a . Generating Particle Beams of Controlled Dimensions and Divergence. 1. Theory of Particle Motion in Aerodynamic Lenses and Nozzle Expansions . Aerosol Sci. Technol. , 22 : 293 – 313 .
- Liu , P. , Ziemann , P. J. , Kittelson , D. B. and McMurry , P. H. 1995b . Generating Particle Beams of Controlled Dimensions and Divergence. 2. Experimental Evaluation of Particle Motion in Aerodynamic Lenses and Nozzle Expansions . Aerosol Sci. Technol. , 22 : 314 – 324 .
- Maria , S. F. , Russell , L. M. , Gilles , M. K. and Myneni , S. C. B. 2004 . Organic Aerosol Growth Mechanisms and Their Climate-Forcing Implications . Science , 306 : 1921 – 1924 .
- Marr , L. C. , Dzepina , K. , Jimenez , J. L. , Reisen , F. , Bethel , H. L. , Arey , J. , Gaffney , J. S. , Marley , N. A. , Molina , L. T. and Molina , M. J. 2006 . Sources and Transformations of Particle-Bound Polycyclic Aromatic Hydrocarbons in Mexico City . Atmos. Chem. Phys. , 6 : 1733 – 1745 .
- Mills , N. L. , Tornqvist , H. , Robinson , S. D. , Gonzalez , M. , Darnley , K. , MacNee , W. , Boon , N. A. , Donaldson , K. , Blomberg , A. , Sandstrom , T. and Newby , D. E. 2005 . Diesel Exhaust Inhalation Causes Vascular Dysfunction and Impaired Endogenous Fibrinolysis . Circulation , 112 : 3930 – 3936 .
- Mitschke , S. , Adam , T. , Streibel , T. , Baker , R. R. and Zimmermann , R. 2005 . Application of Time-of-Flight Mass Spectrometry with Laser-Based Photoionization Methods for Time-Resolved On-Line Analysis of Mainstream Cigarette Smoke . Anal. Chem. , 77 : 2288 – 2296 .
- Murphy , D. M. 2007 . The Design of Single Particle Laser Mass Spectrometers . Mass Spectrom. Rev. , 26 : 150 – 165 .
- Nash , D. G. , Baer , T. and Johnston , M. V. 2006 . Aerosol Mass Spectrometry: An Introductory Review . Int. J. Mass Spectrom. , 258 : 2 – 12 .
- Ning , Z. , Moore , K. F. , Polidori , A. and Sioutas , C. 2006 . Field Validation of the New Miniature Versatile Aerosol Concentration Enrichment System (Mvaces) . Aerosol Sci. Technol. , 40 : 1098 – 1110 .
- Oktem , B. , Tolocka , M. P. and Johnston , M. V. 2004 . On-Line Analysis of Organic Components in Fine and Ultrafine Particles by Photoionization Aerosol Mass Spectrometry . Anal. Chem. , 76 : 253 – 261 .
- Oktem , B. , Tolocka , M. P. , Zhao , B. , Wang , H. and Johnston , M. V. 2005 . Chemical Species Associated with the Early Stage of Soot Growth in a Laminar Premixed Ethylene-Oxygen-Argon Flame . Combust. Flame , 142 : 364 – 373 .
- Park , S. S. , Harrison , D. , Pancras , J. P. and Ondov , J. M. 2005 . Highly Time-Resolved Organic and Elemental Carbon Measurements at the Baltimore Supersite in 2002 . J. Geophys. Res. , : 110
- Robinson , A. L. , Donahue , N. M. and Rogge , W. F. 2006a . Photochemical Oxidation and Changes in Molecular Composition of Organic Aerosol in the Regional Context . J. Geophys. Res. , : 111 (D03302), doi: 10.1029/ 2005/JD006265
- Robinson , A. L. , Donahue , N. M. , Shrivastava , M. K. , Weitkamp , E. A. , Sage , A. M. , Grieshop , A. P. , Lane , T. E. , Pierce , J. R. and Pandis , S. N. 2007 . Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging . Science , 315 : 1259 – 1262 .
- Robinson , A. L. , Subramanian , R. , Donahue , N. M. , Bernardo-Bricker , A. and Rogge , W. F. 2006b . Source Apportionment of Molecular Markers and Organic Aerosol-1. Polycyclic Aromatic Hydrocarbons and Methodology for Data Visualization . Environ. Sci. Technol. , 40 : 7803 – 7810 .
- Robinson , A. L. , Subramanian , R. , Donahue , N. M. , Bernardo-Bricker , A. and Rogge , W. F. 2006c . Source Apportionment of Molecular Markers and Organic Aerosol. 3. Food Cooking Emissions . Environ. Sci. Technol. , 40 : 7820 – 7827 .
- Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. and Cass , G. R. 1998 . Sources of Fine Organic Aerosol. 9. Pine, Oak, and Synthetic Log Combustion in Residential Fireplaces . Environ. Sci. Technol. , 32 : 13 – 22 .
- Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. , Cass , G. R. and Simoneit , B. R. T. 1993a . Sources of Fine Organic Aerosol. 2. Noncatalyst and Catalyst-Equipped Automobiles and Heavy-Duty Diesel Trucks . Environ. Sci. Technol. , 27 : 636 – 651 .
- Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. , Cass , G. R. and Simoneit , B. R. T. 1993b . Sources of Fine Organic Aerosol. 3. Road Dust, Tire Debris, and Organometallic Brake Lining Dust-Roads as Sources and Sinks . Environ. Sci. Technol. , 27 : 1892 – 1904 .
- Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. , Cass , G. R. and Simoneit , B. R. T. 1993c . Sources of Fine Organic Aerosol. 4. Particulate Abrasion Products from Leaf Surfaces of Urban Plants . Environ. Sci. Technol. , 27 : 2700 – 2711 .
- Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. , Cass , G. R. and Simoneit , B. R. T. 1997 . Sources of Fine Organic Aerosol. 7. Hot Asphalt Roofing Tar Pot Fumes . Environ. Sci. Technol. , 31 : 2726 – 2730 .
- Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. , Cass , G. R. and Simonelt , B. R. T. 1991 . Sources of Fine Organic Aerosol. 1. Charbroilers and Meat Cooking Operations . Environ. Sci. Technol. , 25 : 1112 – 1125 .
- Samet , J. M. , Dominici , F. , Curriero , F. C. , Coursac , I. and Zeger , S. L. 2000 . Fine Particulate Air Pollution and Mortality in 20 U.S. Cities, 1987–1994 . N. Engl. J. Med. , 343 : 1742 – 1749 .
- Schauer , J. J. , Kleeman , M. J. , Cass , G. R. and Simoneit , B. R. T. 1999a . Measurement of Emissions from Air Pollution Sources. 1. C1 through C29 Organic Compounds from Meat Charbroiling . Environ. Sci. Technol. , 33 : 1566 – 1577 .
- Schauer , J. J. , Kleeman , M. J. , Cass , G. R. and Simoneit , B. R. T. 1999b . Measurement of Emissions from Air Pollution Sources. 2. C1 through C30 Organic Compounds from Medium Duty Diesel Trucks . Environ. Sci. Technol. , 33 : 1578 – 1587 .
- Schauer , J. J. , Kleeman , M. J. , Cass , G. R. and Simoneit , B. R. T. 2001 . Measurement of Emissions from Air Pollution Sources. 3. C1-C29 Organic Compounds from Fireplace Combustion of Wood . Environ. Sci. Technol. , 35 : 1716 – 1728 .
- Schauer , J. J. , Rogge , W. F. , Hildemann , L. M. , Mazurek , M. A. , Cass , G. R. and Simoneit , B. R. T. 1996 . Source Apportionment of Airborne Particulate Matter Using Organic Compounds as Tracers . Atmos. Environ. , 30 : 3837 – 3855 .
- Simoneit , B. R. T. , Kobayashi , M. , Mochida , M. , Kawamura , K. and Huebert , B. J. 2004 . Aerosol Particles Collected on Aircraft Flights over the Northwestern Pacific Region During the Ace-Asia Campaign: Composition and Major Sources of the Organic Compounds . J. Geophys. Res. , : 109
- Simoneit , B. R. T. , Medeiros , P. M. and Didyk , B. M. 2005 . Combustion Products of Plastics as Indicators for Refuse Burning in the Atmosphere . Environ. Sci. Technol. , 39 : 6961 – 6970 .
- Simoneit , B. R. T. , Schauer , J. J. , Nolte , C. G. , Oros , D. R. , Elias , V. O. , Fraser , M. P. , Rogge , W. F. and Cass , G. R. 1999 . Levoglucosan, a Tracer for Cellulose in Biomass Burning and Atmospheric Particles . Atmos. Environ. , 33 : 173 – 182 .
- Sisler , J. F. and Malm , W. C. 1994 . The Relative Importance of Soluble Aerosols to Spatial and Seasonal Trends of Impaired Visibility in the United States . Atmos. Environ. , 28 : 851 – 862 .
- Stales , C. A. , Peterson , D. R. , Parkerton , T. F. and Adams , W. J. 1997 . The Environmental Fate of Phthalate Esters: A Literature Review . Chemosphere , 35 : 667 – 749 .
- Stephanou , E. G. 2005 . The Decay of Organic Aerosols . Nature , 434 : 31
- Streibel , T. , Weh , J. , Mitschke , S. and Zimmermann , R. 2006 . Thermal Desorption/Pyrolysis Coupled with Photoionization Time-of-Flight Mass Spectrometry for the Analysis of Molecular Organic Compounds and Oligomeric and Polymeric Fractions in Urban Particulate Matter . Anal. Chem. , 78 : 5354 – 5361 .
- Subramanian , R. , Donahue , N. M. , Bernardo-Bricker , A. , Rogge , W. F. and Robinson , A. L. 2006 . Contribution of Motor Vehicle Emissions to Organic Carbon and Fine Particle Mass in Pittsburgh, Pennsylvania: Effects of Varying Source Profiles and Seasonal Trends in Ambient Marker Concentrations . Atmos. Environ. , 40 : 8002 – 8019 .
- Thuren , A. and Larsson , P. 1990 . Phthalate Esters in the Swedish Atmosphere . Environ. Sci. Technol. , 24 : 554 – 559 .
- Tolocka , M. P. , Heaton , K. J. , Dreyfus , M. A. , Wang , S. Y. , Zordan , C. A. , Saul , T. D. and Johnston , M. V. 2006 . Chemistry of Particle Inception and Growth During Alpha-Pinene Ozonolysis . Environ. Sci. Technol. , 40 : 1843 – 1848 .
- Turpin , B. J. , Saxena , P. and Andrews , E. 2000 . Measuring and Simulating Particulate Organics in the Atmosphere: Problems and Prospects . Atmos. Environ. , 34 : 2983 – 3013 .
- Van Bramer , S. E. and Johnston , M. V. 1990 . 10.5-Ev Photoionization Mass Spectrometry of Aliphatic Compounds . J. Am. Soc. Mass Spectrom. , 1 : 419 – 426 .
- Williams , B. J. , Goldstein , A. H. , Kreisberg , N. M. and Hering , S. V. 2006 . An In-Situ Instrument for Speciated Organic Composition of Atmospheric Aerosols: Thermal Desorption Aerosol Gc/Ms-Fid (Tag) . Aerosol Sci. Technol. , 40 : 627 – 638 .
- Williams , B. J. , Goldstein , A. H. , Millet , D. B. , Holzinger , R. , Kreisberg , N. M. , Hering , S. V. , White , A. B. W. , Douglas , R. , Allan , J. D. and Jimenez , J. L. 2007 . Chemical Speciation of Organic Aerosol During the International Consortium for Atmospheric Research on Transport and Transformation 2004: Results from in Situ Measurements . J. Geophys. Res. , : 112 (010826), doi: 10.1029/2006JD007601
- Zanobetti , A. and Schwartz , J. 2006 . Air Pollution and Emergency Admissions in Boston, MA . J. Epidemiol. Community Health , 60 : 890 – 895 .
- Zhang , Q. , Alfarra , M. R. , Worsnop , D. R. , Allan , J. D. , Coe , H. , Canagaratna , M. R. and Jimenez , J. L. 2005a . Deconvolution and Quantification of Hydrocarbon-Like Oxygenated Organic Aerosols Based on Aerosol Mass Spectrometry . Environ. Sci. Tech. , 39 : 4938 – 4952 .
- Zhang , Q. , Worsnop , D. R. , Canagaratna , M. R. and Jimenez , J. L. 2005b . Hydrocarbon-Like and Oxygenated Organic Aerosols in Pittsburgh: Insights into Sources and Processes of Organic Aerosols . Atmos. Chem. Phys. Discuss. , 5 : 8421 – 8471 .