Abstract
Accurately predicting formation and partitioning of ambient organic aerosols remains a challenge despite decades of sustained effort in this domain. A major source of uncertainty is the poorly characterized volatility of these aerosols. This uncertainty stems in large part from difficulty separating the overlapping effects of aerosol thermodynamic properties and evaporation coefficients in thermodenuder volatility studies. For lack of other information, it is commonly assumed that the evaporation coefficient is unity when interpreting thermodenuder data, leading to potentially large biases in inferred volatility of the sampled aerosol. In this paper, we present a novel thermodenuder-based approach for determining evaporation coefficients of pure compound and complex aerosols without knowledge of their thermodynamic properties. The method involves tracing the normalized dynamic response of an aerosol system to a step change in temperature as it flows through a heated tube. The approach is validated using pure compounds and a mixture of laboratory-generated dicarboxylic acids, and is applied to concentrated ambient aerosols sampled in Beirut, Lebanon. Three valid data sets were obtained from more than 200 h of ambient air sampling during the month of August 2010, yielding values of 0.34, 0.46, and 0.28 for an assumed binary gas diffusion coefficient of 7.8 × 10−6 m2/s at 60°C.
Copyright 2012 American Association for Aerosol Research
1. INTRODUCTION
Semivolatile organic compounds constitute a major fraction of both primary and secondary organic aerosols (SOA), and their presence in the atmosphere contributes greatly to uncertainty in predicting atmospheric aerosol concentrations (Donahue et al. Citation2006). A major factor contributing to this uncertainty is the lack of knowledge of their basic thermodynamic properties, particularly their volatility (Pankow and Barsanti Citation2009). Understanding the volatility of semivolatile organic compounds is thus central to improving atmospheric aerosol models. One widely used technique to estimate aerosol particle volatility is to infer it from change in particle volume concentration after drawing the aerosol through a temperature-controlled flow tube, or thermodenuder (TD; Wehner et al. Citation2002; An et al. Citation2007; Huffmann et al. Citation2008; Saleh et al. Citation2008; Faulhaber et al. Citation2009; Dzepina et al. Citation2009; Cappa and Jimenez Citation2010).
However, because time scales of evaporation are often of the order of the residence times available in practical TDs, one of the main challenges of TD-based volatility measurements has been accounting for the overlapping effects of thermodynamic and kinetic properties of the aerosol, namely the diffusion coefficient (D), and the evaporation coefficient (α), when interpreting the measurements. While D can be estimated with reasonable confidence from molecular structure (e.g., Wilke and Lee Citation1955; Bird et al. Citation1960; Reid et al. Citation1987), α is usually unknown. As a result, most workers in this domain have resorted to assuming an evaporation coefficient of unity, leading to potentially large biases in reported thermodynamic properties (Saleh et al. Citation2009). Recognizing this limitation, Cappa and Jimenez Citation(2010) recently reported TD measurements of ambient aerosols, and provided estimates for volatility distributions over a range of assumed values of α. They showed that the inferred volatilities were highly sensitive to α.
There have been several attempts to estimate evaporation coefficients of semivolatile organic aerosols. Since α has a direct effect on the rate of evaporation of aerosol particles, it is intuitive to attempt to estimate it from measurements of evaporation kinetics. However, as mentioned above, the main difficulty is that the effect of α on evaporation kinetics overlaps with that of the volatility of the evaporating species. Due to this difficulty, some of the previous studies have aimed to “constrain” α rather than obtain a direct measurement. For example, Stanier et al. (2007) studied evaporation of SOA generated by α-pinene ozonolysis. They estimated that α < 0.1 was required to interpret evaporation rates in tandem differential mobility analysis (TDMA) experiments. On the other hand, Cappa and Jimenez Citation(2010) found that α > 0.01 was needed to explain observed evaporation of ambient aerosols in a TD. To our knowledge, the only study that decoupled the effect of α and volatility of semivolatile organic aerosols was performed by our group using integrated volume—tandem differential mobility analysis (IV-TDMA; Saleh et al. Citation2009). In this approach, volatility (saturation pressure and enthalpy of vaporization) is first determined using the integrated volume method (IVM), an equilibrium-based method requiring no knowledge of α. Then, α is determined from single-parameter optimization of an evaporation kinetics model in which the objective is to minimize difference with observed evaporation in TDMA experiments. Values of α were found to be of O(10−1) for lab-generated pure succinic, adipic, and pimelic acid aerosols. While we have shown that the IV-TDMA can be used to determine α of pure semivolatile aerosols, extension to complex aerosol systems (e.g., smog chamber and ambient aerosols) is not straightforward. There are large uncertainties in derived effective thermodynamic properties of semivolatile organics (see, for example, Cappa and Jimenez Citation2010) using current lumping techniques (e.g., Odum et al. Citation1997; Donahue et al. Citation2006), which make it difficult to obtain an estimate of α from TDMA measurements.
TABLE 1 Properties of the volatility bins used in the theoretical experiments
In this paper, we present a novel method to determine α of an aerosol from measurements of equilibration profiles in a TD. We have previously shown that equilibration time of an aerosol flowing in a TD is not a function of thermodynamic properties, and is governed solely by the aerosol size distribution and kinetic properties, namely α and D (Saleh et al. Citation2011). Here, we validate this method by using it to estimate α of lab-generated pure dicarboxylic acids and comparing the results to those obtained with the IV-TDMA (Saleh et al. Citation2009). Extension of the method to multicomponent aerosol is tested by applying it to a lab-generated mixture of dicarboxylic acids. Finally, we illustrate how this method can be applied to complex aerosol systems by using it to determine effective α of ambient aerosols in Beirut, Lebanon, the first time such a determination has been made to our knowledge. As discussed in Saleh et al. Citation(2009), α determined here is an effective value, which lumps all processes underlying the particle–gas exchange. For example, for sublimation from a solid surface, α may include intrinsic surface rearrangement and dissociation kinetics, as well as finite rate diffusion from the interior to the surface.
2. THEORY
2.1. Approach
When perturbed from phase equilibrium by a change in temperature or pressure, an aerosol experiences phase change in the direction needed to restore equilibrium, i.e., toward a vapor phase saturation ratio, Cg* = Cg/Csat (T), of unity. Provided that sufficient particle mass is available for equilibration at the new condition, the evolution of Cg* as the aerosol returns to equilibrium—the “equilibration profile”—is governed strictly by the particle size distribution and kinetic properties of the aerosol, in particular the diffusion coefficient, D, and evaporation coefficient, α (Saleh et al. Citation2011). Thus, given the size distribution and D, the evaporation coefficient can, in principle, be inferred from the shape of an experimentally measured equilibration profile, Cg*(x), without reference to thermodynamics.
Here, we apply this principle to a polydisperse aerosol experiencing a quasi-step change in temperature as it flows through a heated tube, or “thermodenuder.” At various distances from the tube inlet, the change in particle volume concentration relative to the inlet, ΔV(x), is measured, e.g., by integrating the volume distribution obtained using an scanning mobility particle spectrometer (SMPS). ΔV(x) initially increases monotonically with distance from the inlet, and then approximately levels off at some equilibrium value, ΔV equil. The equilibrium ΔV represents the amount of particle volume required to evaporate in order to bring about equilibrium with the vapor phase at the elevated temperature. If the saturation pressure of the aerosol at its initial state is orders of magnitude smaller than the saturation pressure at the higher temperature of the TD, then Cg*(x) can be determined directly from measurements of ΔV using Equation (1), i.e.,
For the current study, the TD temperature was 60°C, and the initial aerosol temperature was approximately 25°C. For ΔH > 100 kJ/mol, which is realistic for laboratory-generated (Saleh et al. Citation2008, 2009, 2010) and ambient (Cappa and Jimenez Citation2010; Epstein et al. Citation2010) semivolatile organics, ![]() . Equation (1) can therefore be used with good accuracy to measure an equilibration profile based on particle volume measurements.
. Equation (1) can therefore be used with good accuracy to measure an equilibration profile based on particle volume measurements.
Given the measured Cg*(x), the evaporation coefficient can be obtained by fitting the governing kinetic equation to the data. We have previously shown (Saleh et al. Citation2011) that the equilibration profile of an aerosol undergoing phase change is governed by
Here, t* = t/tr
is dimensionless time, where tr
is the residence time in the TD. dp* =dp/dp, in
is dimensionless particle size, where dp,
in is the initial particle diameter. N
tot is the total particle number concentration, K is the Kelvin correction for curvature effect, and D is the binary diffusion coefficient. ![]() is the Fuchs–Sutugin correction for noncontinuum effects (Fuchs and Sutugin Citation1971). For an evaporating aerosol with approximately constant particle size, Equation (2) represents a first-order system with a characteristic time constant τ=1/2πN
tot
d
p,inDF, the time required to bring the gas phase concentration to Cg* = 1/e. In the case of polydisperse aerosols, the above diameters are represented by the condensation sink diameter given by Lehtinen et al. Citation(2003).
is the Fuchs–Sutugin correction for noncontinuum effects (Fuchs and Sutugin Citation1971). For an evaporating aerosol with approximately constant particle size, Equation (2) represents a first-order system with a characteristic time constant τ=1/2πN
tot
d
p,inDF, the time required to bring the gas phase concentration to Cg* = 1/e. In the case of polydisperse aerosols, the above diameters are represented by the condensation sink diameter given by Lehtinen et al. Citation(2003).
Since the aerosol distribution (N tot, dp, in) can be measured and D can be estimated from semiempirical correlations (e.g., Reid et al. Citation1987), a single-parameter optimization can be performed on Equation (2) to determine the value of α that fits an experimental equilibration profile.
In sum, the method is analogous to determining the time constant from a step response of a linear first-order dynamic system.
2.2. Multicomponent Aerosol
This approach can be extended to mixtures of compounds through the use of an effective saturation concentration and evaporation coefficient for the mixture. For an ideal solution, the mixture effective saturation concentration can be represented as the average of the saturation concentrations of the individual components C sat,i weighted by the corresponding mole fractions (xi ): C sat,eff=∑xiC sat,i . Similarly, the gas–phase mixture concentration, Cg, eff, can be written as C g,eff=∑C g,i . Assuming K of unity, Equation (2) can therefore be written for the mixture as
Here, F eff is the effective Fuchs–Sutugin correction for the mixture. By comparing Equations (2) and (3), it can be readily shown that
Since at equilibrium, C sat,eff=C g,eff=∑xiC sat,i , Equation (4) can be solved for the effective evaporation coefficient, α eff, of the mixture
where A = 1 + Kn, B = 1 + 0.3773Kn, and C = 1.33Kn(1 + Kn). Equation (5) expresses a physically intuitive result that the effective evaporation coefficient is an evaporation rate-weighted average of the individual component evaporation coefficients. In general, the detailed composition, thermodynamic properties, and evaporation coefficients are unknowns, so the real value of Equation (5) is to simply illustrate that the effective evaporation coefficient derived using an effective mixture C sat is physically grounded.
Consider, for example, a theoretical experiment in which an internally mixed, monodisperse aerosol flows through a heated tube. The 100 nm diameter aerosol has a total initial concentration (C 0) of 350 μg/m3 and is distributed into four volatility bins with properties given in . The mixture is further assumed to form an ideal solution. The evolution of the mass concentration of each bin in the aerosol is numerically simulated using the plug flow model described in Saleh et al. Citation(2008). A no-flux boundary condition is imposed at the tube wall.
shows the change of mass concentration of each bin in the particle phase (ΔCi ), which is equal to the vapor build-up of each component in the gas phase (ΔCg,i ). also shows the evolution of the total change in aerosol concentration (ΔC = ∑ΔCi ). Treating the simulated ΔC as the output of an experiment, the volatility profile Cg* (t) can be calculated using Equation (1). As described in Section 2.1 above, Equation (2) is then optimized to determine α eff. We stress that in optimizing Equation (2) for αeff, no information is used about the volatility distribution, the corresponding mole fractions, or α value of each bin. The only information used is ΔC, as would be the case in a live experiment.
FIG. 1 Simulated change in particle mass concentration of each component of a four-component hypothetical aerosol mixture in a TD (theoretical experiment 1). C 0 = 350 μg/m3, d 0 = 100 nm, T 0 = 25°C, and T TD = 60°C. C sat values are given at 25°C.
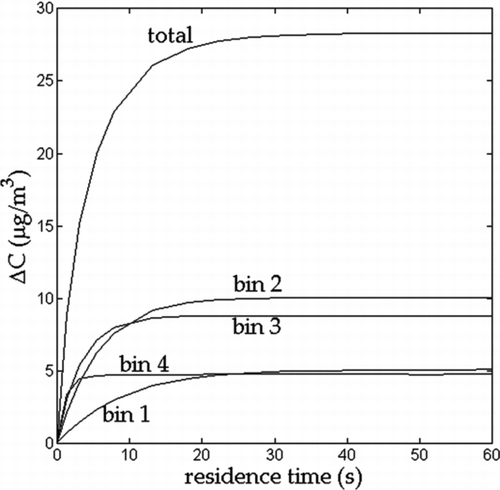
The results are shown in . The α eff obtained by fit to Equation (2) was 0.19, which is of the same value as given by Equation (5), using the initial mole fractions. We have performed several similar simulations and found that the agreement between the fitted α eff and the value predicted by Equation (5) remains exact regardless of the volatility distribution or component α i 's, provided that the mole fractions in the particle phase do not change greatly throughout the evaporation process. This is equivalent to assuming that the fractional change in aerosol mass concentration is small during the equilibration process.
FIG. 2 Equilibration profiles of the simulated aerosol mixture obtained from change in total particle concentration using Equation (2) (diamonds), and from optimization of Equation (1) with the whole mixture represented by an effective single C sat (lines).
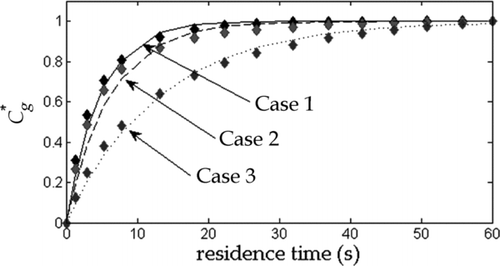
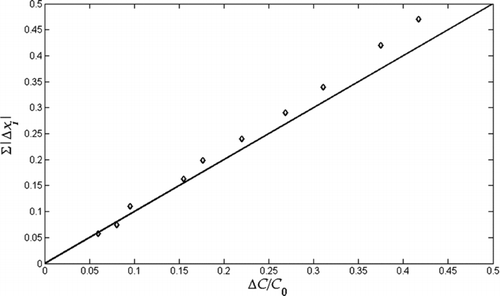
FIG. 3 Simulated change in absolute mole fractions (calculated as the sum of the absolute change for each volatility bin, ∑|Δxi |) versus mass fraction evaporated for a wide range of model aerosols. Properties used for these simulations are given in Table 1.
If the mole fractions do change significantly upon evaporation in the TD, α
eff will deviate from the initial value and will be a function of time: ![]() . The value of C
sat,eff will also become time-dependent: C
sat,eff(t)=∑xi
(t)C
sat,i
. Thus, the equilibration profile will depart from the first-order exponential decay given by Equation (2). To explore the implications of this departure on α
eff, we performed simulations with varying volatility distributions (mole fractions), evaporation coefficients, and aerosol loadings. The ranges of the variables used in the simulations are given in . Changes in mole fractions during evaporation in the TD were computed as the sum of the absolute change of the mole fraction in each volatility bin (∑|Δxi
|). We found that ∑|Δxi
| is linearly correlated with ΔC/C
0, or C
sat,eff/C0
, and the relation almost follows a 1-to-1 line as shown in . Thus, for experiments where xi
are not known, ΔC/C
0, which is the measured quantity, can be used to estimate the change in mole fractions. shows the error in the measured α
eff (the deviation from theoretical α
eff) plotted versus ΔC/C. For ΔC/C
0 < 0.4, the error in measured α
eff is less than 25%. In practice, ΔC/C
0 can be minimized by increasing C
0 or decreasing the TD temperature.
. The value of C
sat,eff will also become time-dependent: C
sat,eff(t)=∑xi
(t)C
sat,i
. Thus, the equilibration profile will depart from the first-order exponential decay given by Equation (2). To explore the implications of this departure on α
eff, we performed simulations with varying volatility distributions (mole fractions), evaporation coefficients, and aerosol loadings. The ranges of the variables used in the simulations are given in . Changes in mole fractions during evaporation in the TD were computed as the sum of the absolute change of the mole fraction in each volatility bin (∑|Δxi
|). We found that ∑|Δxi
| is linearly correlated with ΔC/C
0, or C
sat,eff/C0
, and the relation almost follows a 1-to-1 line as shown in . Thus, for experiments where xi
are not known, ΔC/C
0, which is the measured quantity, can be used to estimate the change in mole fractions. shows the error in the measured α
eff (the deviation from theoretical α
eff) plotted versus ΔC/C. For ΔC/C
0 < 0.4, the error in measured α
eff is less than 25%. In practice, ΔC/C
0 can be minimized by increasing C
0 or decreasing the TD temperature.
FIG. 4 Predicted error in measured α eff relative to theoretical α eff versus mass fraction evaporated for a range of model aerosols. Properties used for these simulations are given in Table 1.
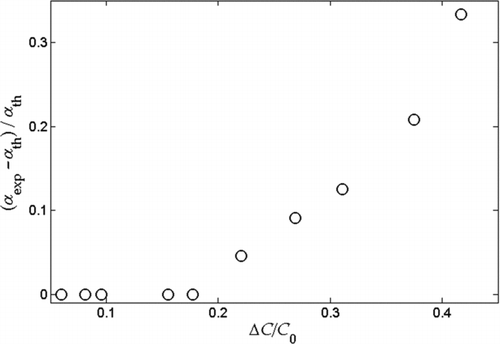
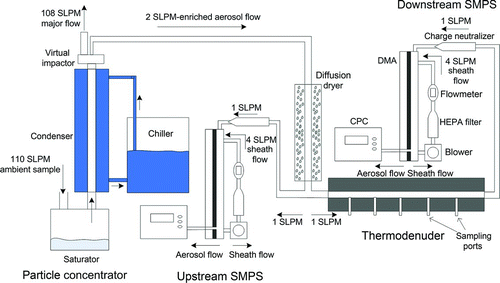
FIG. 5 Schematic of experimental setup. (Color figure available online.)
In summary, the results of these simulations show that in the absence of detailed information about aerosol composition in complex mixtures, it is possible to estimate an effective α from the overall equilibration profile based on an effective mixture C sat.
3. EXPERIMENTAL
The experimental setup, shown in , had two configurations: one for measurement of laboratory-generated aerosols, and one for ambient aerosols. The former setup is described in detail in Saleh et al. Citation(2011). Briefly, the TD was 1 m long and 2.5 cm ID, which provided an average residence time of approximately 30 s at 1 LPM. Residence times of 20, 15, 12, and 9 s were achieved by changing the flow rate to 1.5, 2, 2.5, and 3.5 LPM respectively. For ambient aerosol measurements, the setup included a particle enrichment system and a different TD design. The TD was 1 m long and 3.5 cm ID, providing a maximum residence time of 58 s at 1 LPM. The TD was fitted with extraction ports at 17, 34, 52, 69, and 87 cm, which provided average residence times of 10, 20, 30, 40, and 50 s at 1 LPM. The TD was maintained at a temperature of 40°C for laboratory aerosols and 60°C for ambient aerosol measurements. For both TD designs, particle deposition was found to be negligible.
Integrated particle volume was obtained using daily intercalibrated SMPS systems (TSI 3081 DMA or equivalent, TSI 3772 CPCs) scanning in the size range 12–650 nm. The change in volume, ΔV(t), of the concentrated ambient aerosol upon heating in the TD was obtained as the difference between integrated particle volume measured upstream of the TD and at each extraction port. Integrated volume at each port was determined from the mean of 15 measurements using an SMPS scan time of 90 s. The time required to obtain a complete data set (i.e., 15 scans × 5 ports) was approximately 3 h. This could be shortened considerably by executing the measurements simultaneously across the five ports using multiple SMPS systems operating in parallel.
3.1. Ambient Aerosol Sampling
As evident in the definition of the characteristic time constant τ in Equation (2), the equilibration profile of a given aerosol depends on the particle size distribution, specifically the aerosol particle length, N tot dp . To obtain a valid experimental equilibration profile, the aerosol length must be constant for the duration of the experiment. While for lab-generated aerosols, the particle size distribution can be held constant, when performing measurements on ambient aerosols fluctuations in ambient concentration constitute a major challenge. We addressed this by using the laboratory space as a volume damper. Outside air was continuously drawn into the 95 m3 room from a 1.5 m2 window at approximately 0.1 air changes per h for the duration of the campaign. There was no air supply source other than the window, and there was no activity inside the laboratory during the measurements other than the operating setup. While these measures helped eliminate high-frequency fluctuations, low-frequency drift in total concentration remained. Consequently, more than 200 h of sampling over a period of 3 weeks yielded only three reliable data sets, all occurring between the hours of 1 AM and 5 AM.
Use of multiple SMPS systems operating in parallel on each TD port would have reduced the measurement window during which the ambient aerosol was required to remain stable from 3 h to several minutes, and would likely have permitted acquisition of many more valid data sets during the campaign.
3.2. Ambient Particle Enrichment
We have previously found that ambient particulate matter (PM) concentrations are too low to achieve equilibrium in a practical TD design (Saleh et al. Citation2011). To obtain experimental equilibration profiles, we therefore concentrated the ambient PM using an aerosol particle concentrator based on the design of Sioutas et al. Citation(1999). Briefly, particle concentration was achieved in a virtual impactor with cutoff size of 1.5 μm at an inlet flow rate of 110 LPM. Upstream of the virtual impactor, submicrometer ambient particles were grown to supermicrometer sizes via water condensation in a saturator/condenser system.
As shown in , ambient air flowing at 110 LPM is first drawn through the saturator, a vessel containing de-ionized water maintained at 33–35°C. The average residence time in the saturator is 3 s, which is sufficient to achieve saturation (Sioutas et al. Citation1999). The sample then passes through the condenser, where the particles are grown by heterogeneous condensation of water vapor. The condenser is a counterflow single-pass shell and tube heat exchanger. On the tube side, the aerosol flows through a 1 m long 2.5 cm ID stainless steel tube with an average residence time of approximately 0.3 s at 110 LPM. The jacket is an insulated polycarbonate cylinder 1 m long × 7.5 cm ID. A water/antifreeze mixture maintained at −5°C circulates through the jacket. Circulation is provided by an automatically controlled chiller (Julabo, model F33).
The flow exiting the condenser is then drawn through a virtual impactor operating with a minor flow rate of 2 LPM. Excess moisture is removed from the aerosol-enriched 2 LPM flow in a diffusion dryer (TSI model 3062). The enrichment ratio achieved with this setup was 30–35, on a number basis.
3.3. Site Description
Ambient measurements were conducted during the month of August 2010 at the American University of Beirut, a green campus located along the Mediterranean seafront in the urban core of Beirut, Lebanon. The sampled air was taken at an elevation 25 m above ground level, with the nearest urban boulevard located approximately 160 m north of the site. Summer months in Beirut are essentially precipitation-free and characterized by a largely constant visible haze over the city. While the picture is complicated by sea breeze phenomena, seasonal and diurnal features in measured ambient CO, O3, PM, and C1–C3 carbonyl compounds are consistent with vehicular emissions as the major source of primary ambient aerosols, and significant subsequent photochemical processing and formation of secondary organic aerosols (Moussa et al. Citation2006; Saliba et al. Citation2006). Results presented in the current study are for particles sampled between 1 AM and 5 AM, during which urban activity is minimal, suggesting that the sample aerosols were relatively aged and contained a significant proportion of secondary aerosols. Ambient temperature during these hours was approximately steady at 28°C and the relative humidity was 74–79%.
3.4. Uncertainty Analysis
We used a Monte Carlo approach to assess the propagation of measurement uncertainty to α values obtained by optimization of Equation (2). The mean and standard deviation of ΔV at each port were calculated from the 15 measurements, from which the mean experimental Cg* and standard deviation at each port were calculated. One hundred simulations were performed with perturbed values of experimental Cg* defined as ![]() + φσ, where σ is the standard deviation and φ a random variable drawn from a normal distribution with mean of 0 and standard deviation of 1. Results from the simulations were used to calculate mean values and uncertainties (95% confidence interval) in α.
+ φσ, where σ is the standard deviation and φ a random variable drawn from a normal distribution with mean of 0 and standard deviation of 1. Results from the simulations were used to calculate mean values and uncertainties (95% confidence interval) in α.
4. RESULTS AND DISCUSSION
4.1. Evaporation Coefficients of Laboratory-Generated Aerosols
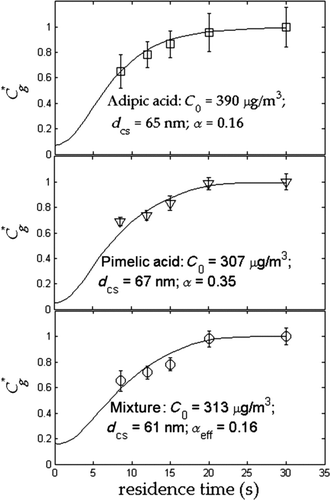
Equilibration profiles were obtained from previously measured experimental data (Saleh et al. Citation2011) for three lab-generated dicarboxylic acid aerosol systems, namely 1) adipic acid, 2) pimelic acid, and 3) a mixture of succinic, adipic, pimelic, and azelaic acids. Systems 1 and 2 illustrate the determination of α of pure compounds, while system 3 illustrates the determination of an effective α of a multicomponent mixture of semivolatile compounds. Experimental equilibration profiles calculated using Equation (1) are shown in . Also shown are theoretical equilibration profiles obtained from single-parameter (α) optimization of Equation (2). The values of D at TD temperature of 40°C used in Equation (2) were calculated from a semiempirical correlation (Reid et al. Citation1987) as 7.6 ×10−6, 6.5 ×10−6, and 6.6 ×10−6 m2/s for systems 1, 2, and 3, respectively. For system 3, D was estimated as a mass-weighted average of the components.
FIG. 6 Measured and simulated dimensionless vapor build-up profiles from Saleh et al. Citation(2011). T in = 25°C and T TD = 40°C. Mixture consists of succinic, adipic, pimelic, and azelaic acid in respective mole fractions of 0.1, 0.4, 0.4, and 0.1. Error bars correspond to 95% confidence intervals calculated using two-tailed Student's-t distribution; N = 10–15 for each data point.
Evaporation coefficient values obtained for the three systems are given in , along with values obtained previously using the IV-TDMA method (Saleh et al. Citation2009). (We note that IV-TDMA values reported in Saleh et al. Citation(2009) have been modified from the original publication to reflect corrected values of Knudsen number used in the Fuchs–Sutugin expression. As originally reported, the α values given in Saleh et al. Citation(2009) were based on Knudsen numbers computed using the mean free path of air at 25°C, whereas the corrected values reported in use mean free path of the vapor species at the thermodenuder temperature, and are thus more accurate. The previously reported values were low by approximately 30%).
TABLE 2 Evaporation coefficients of lab-generated and ambient aerosols
IV-TDMA utilizes a kinetic model to fit α to measured particle diameter changes in a near-zero saturation ratio environment; it therefore provides a measure of α which is independent of the method used in the current study. The agreement between the two methods evident in thus confirms empirically the theoretical finding presented in Section 2 that evaporation coefficients can be estimated from experimental equilibration profiles without knowledge of thermodynamic properties.
For the multicomponent case, curve c of shows that an evaporation model based on a single effective mixture α provides a reasonable representation of the evolution of the experimentally measured equilibration profile for a real mixture, even without knowledge of the component properties and their interdependencies. Using Equation (5), we can estimate the ideal solution α eff using C sat values from Saleh et al. (2008, 2009, 2010) and α values from Saleh et al. Citation(2009). Since we do not have an estimate for α of azelaic acid, we assumed a value ranging between 0.1 and 0.3. The theoretical value of α eff ranges between 0.23 and 0.24, which is comparable to the experimentally obtained value of 0.16.
4.2. Evaporation Coefficient of Ambient Aerosol
shows experimental dimensionless equilibration profiles for the three successful ambient data sets. For these measurements, the particle volume concentrations were 160–288 μm3/cm3, and the change in volume concentration upon evaporation in the TD was 25–30 μm3/cm3; thus the relative change in particle concentration (ΔC/C 0) was less than 0.2. Using the analysis in Section 2 and the simulation results in , the deviation from Equation (2) due to change of mole fractions upon evaporation is estimated to be small, and the error associated with α eff is less than 2%. Importantly, it can be seen that the concentrated aerosol achieved equilibration within 40 s, and that, like the laboratory-generated aerosols, the experimental data are well-correlated to a model that describes evaporation using a single effective α.
FIG. 7 Measured and simulated dimensionless equilibration profiles of concentrated ambient aerosol for three different days. T in = 25°C and T TD = 60°C. Samples 2 and 3 measured data are shifted in the plot by 0.5 s to the left and 0.5 s to the right, respectively, for clarity. Error bars correspond to 95% confidence intervals calculated using two-tailed Student's-t distribution; N = 10–15 for each data point.
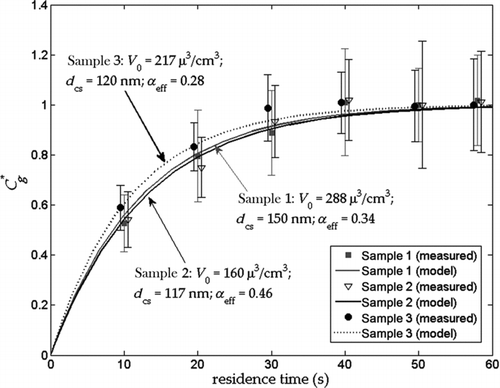
The α values obtained for the three data sets are given in . Although the number of samples is not sufficient to draw general conclusions, these measurements provide first direct evidence that α of ambient urban aerosol is of O(0.1), and of similar magnitude to α reported above for pure and mixed organic acid compounds found in SOA. The measurements are in agreement with the finding of Cappa and Jimenez Citation(2010) that α > 0.01 is required for observed evaporation rates in a TD of ambient aerosols in Mexico City. Cappa and Jimenez Citation(2010) also showed that an assumed α of 0.1 (at a D of 3.5 × 10−6 m2/s) would result in quantitative estimates of gas-phase ambient semivolatile species 30–80% greater than the estimate where α of unity is assumed. Therefore, the current measurements indicate that urban aerosols may be considerably more volatile than previously thought.
It should be noted that in the absence of chemical speciation measurements, the values of α reported here are based on D and molar mass equal to that of pimelic acid (7.8 ×10−6 m2/s at 60°C). A change in assumed D results in an almost directly proportional change in α. A reasonable range of values of D for atmospheric aerosols at 25°C is 3.5 × 10−6 m2/s (Cappa and Jimenez Citation2010) to 1.0 × 10−5 m2/s (Marcolli et al. 2004), which is approximately between 4 × 10−6 m2/s and 1.2 × 10−5 m2/s at 60°C. Based on this range, our estimates of α are bounded by a factor of 2.
Several groups (Virtanen et al. Citation2010; Vaden et al. Citation2011; Cappa and Wilson Citation2011) have recently argued that evaporation of SOA may be rate-limited by diffusional resistance to molecular transport within particles, and have therefore questioned whether quasi-equilibrium partitioning theory is valid for describing evaporation kinetics of SOA. While a review of the strengths and limitations of the evidence for this proposition is beyond the scope of this paper, it merits noting that one advantage of the method demonstrated here is that no assumptions are made regarding the degree to which partitioning theory is applicable; computed fluxes depend on an effective measured saturation pressure exerted at the particle surface, regardless of its underlying determinants (e.g., volatility profiles, activities, diffusion resistances). Further, as discussed in Saleh et al. Citation(2009), our approach lumps particle-phase kinetic constraints into the effective α, and is therefore directly applicable to lumped mass transfer analysis, such as when interpreting thermograms.
We also note that the presence of nonvolatile material in the ambient particles does not affect the analysis presented in this paper. The implications of the presence of nonvolatile component(s) can be explored in terms of one of the following scenarios: 1) If the aerosol is composed of a nonvolatile core and a semivolatile shell, as is usually assumed for black carbon with an organic coating, the nonvolatile component has no effect on the measured evaporation coefficient. 2) If the nonvolatile component is in solution with the semivolatile compounds, it will have no effect on the equilibration profile, as discussed in Saleh et al. Citation(2011). In brief, suppression of evaporation rate due to the presence of the nonvolatile component is counterbalanced by the suppression in equilibration vapor pressure. Thus, there will be no effect on the evaporation coefficient. 3) If the nonvolatile component forms a separate phase that occupies space at the surface of the aerosol particle, it will suppress the evaporation rate (by reducing the surface area), and this effect will actually be factored in the derived evaporation coefficient.
Apart from the small number of successful measurements, the current study is also limited by a potential artifact associated with the aerosol particle enrichment system. It has been previously shown that the concentration of semivolatile substances can be increased in the concentrator by a greater magnitude than the concentration of nonvolatile species (Khlystov et al. Citation2005). This artifact, however, was shown to be relatively minor. Finally, it should be noted that in the use of Equation (1), the density of the ambient aerosol is assumed constant during the evaporation process; this limitation can be readily overcome by use of mass-based concentration measurements.
5. CONCLUSIONS
We have demonstrated a novel technique to determine evaporation coefficients of semivolatile aerosols, as well as effective evaporation coefficients of complex aerosol systems, such as those sampled from smog chambers and ambient air. This technique relies on measurement of aerosol phase equilibration kinetics in a thermodenuder, and does not require knowledge of thermodynamic properties or detailed information of aerosol composition.
We have applied the method to lab-generated pure and mixed dicarboxylic acid aerosols. The measured evaporation coefficients were in excellent agreement with values we have previously obtained using a fundamentally different method. We have also illustrated the use of this method to determine effective evaporation coefficients of complex aerosol systems by applying it to concentrated ambient aerosols in Beirut, Lebanon. For the three successful experiments performed during this study, the obtained values of α were approximately 0.4, indicating that ambient gas phase concentrations of semivolatile species may be considerably greater than assumed in current ambient pollution models.
Acknowledgments
We gratefully acknowledge Professor Constantinos Sioutas of the University of Southern California for helpful discussions and loaning us a virtual impactor for the particle enrichment system. We also gratefully acknowledge the efforts of Mr. Ezzat Jaroudi and Mr. Hassan Beydoun in setting up, calibrating, and trouble-shooting the instrumentation and particle enrichment system used in the study. This work was supported by the University Research Board of the American University of Beirut and the NSF (grant AGS-09-55845).
Related Research Data
REFERENCES
- An , W. J. , Pathak , R. K. , Lee , B. H. and Pandis , S. N. 2007 . Aerosol Volatility Measurement Using an Improved Thermodenuder: Application to Secondary Organic Aerosol . J. Aerosol Sci. , 38 : 305 – 314 .
- Bird , R. B. , Stewart , W. E. and Lightfoot , E. N. 1960 . Transport Phenomena , New York : John Wiley & Sons .
- Cappa , C.D. and Jimenez , J.L. 2010 . Quantitative Estimates of the Volatility of Ambient Organic Aerosol . Atmos. Chem. Phys. , 10 : 5409 – 5424 .
- Cappa , C. D. and Wilson , K. R. 2011 . Evolution of Organic Aerosol Mass Spectra Upon Heating: Implications for OA Phase and Partitioning Behavior . Atmos. Chem. Phys. Discuss. , 11 : 1 – 17 .
- Donahue , N. M. , Robinson , A. L. , Stanier , C. O. and Pandis , S. , N. 2006 . Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics . Environ. Sci. Technol. , 40 : 2635 – 2643 .
- Dzepina , K. , Volkamer , R. M. , Mandronich , S. , Tulet , P. , Ulbrich , I. M. , Zhang , Q. , Cappa , C. D. , Ziemann , P. J. and Jimenez , J. L. 2009 . Evaluation of Recently-Proposed Secondary Organic Aerosol Models for a Case Study in Mexico City . Atmos. Chem. Phys. , 9 : 5681 – 5701 .
- Epstein , S. A. , Riipinen , I. and Donahue , N. M. 2010 . A Semi-Empirical Correlation Between Enthalpy of Vaporization and Saturation Concentration for Organic Aerosol . Environ. Sci. Technol. , 44 : 743 – 748 .
- Faulhaber , A. E , Thomas , B. M. , Jimenez , J. L. , Jayne , J. T. , Worsnop , D. R. and Ziemann , P. J. 2009 . Characterization of Thermodenuder-Particle Beam Mass Spectrometer System for the Study of Organic Aerosol Volatility and Composition . Atmos. Meas. Tech. , 2 : 15 – 31 .
- Fuchs , N. A. and Sutugin , A. G. 1971 . “ High Dispersed Aerosols ” . In Topics in Current Aerosol Research , Edited by: Hidy , G. M. and Brock , J. R. Vol. 2 , 1 – 60 . Oxford : Pergamon Press .
- Huffmann , J. A. , Ziemann , P. J. , Jayne , J. T. , Worsnop , D. R. and Jimenez , J. L. 2008 . Development of Fast-Stepping/Scanning Thermodenuder for Chemically-Resolved Aerosol Volatility Measurements . Aerosol Sci. Technol. , 42 : 395 – 407 .
- Khlystov , A. , Zhang , Q. , Jimenez , J.-L. , Stanier , S. , Pandis , S. , Canagaratna , M. R. , Fine , P. , Misra , C. and Sioutas , C. 2005 . In-Situ Concentration of Semi-Volatile Aerosol Using Water-Condensation Technology . J. Aerosol Sci. , 36 : 866 – 880 .
- Lehtinen , K. J. , Korhonen , H. , Dal Maso , M. and Kulmala , M. 2003 . On the Concept of Condensation Sink Diameter . Boreal Environ. Res. , 8 : 405 – 411 .
- Moussa , S. , El-Fadel , M. and Saliba , N. 2006 . Seasonal, Diurnal and Nocturnal Behaviors of Lower Carbonyl Compounds in the Urban Environment of Beirut, Lebanon . Atmos. Environ. , 40 : 2459 – 2468 .
- Odum , J. R. , Hoffman , T. , Bowman , F. , Collins , D. , Flagan , R. C. and Seinfeld , J. H. 1997 . Gas/Particle Partitioning and Secondary Organic Aerosol Yields . Environ. Sci. Technol. , 30 : 2580 – 2585 .
- Pankow , J. F. and Badsanti , K. C. 2009 . The Carbon Number Polarity Grid: A Means to Manage the Complexity of the Mix of Organic Compounds When Modeling Atmospheric Organic Particular Matter . Atmos. Environ. , 43 : 2829 – 2835 .
- Reid , R. C. , Prausnitz , J. M. and Polling , B. E. 1987 . The Properties of Gases and Liquids , New York : McGraw Hill .
- Saleh , R. , Shihadeh , A. and Khlystov , A. 2009 . Determination of Evaporation Coefficients of Semi-Volatile Aerosols Using an Integrated Volume-Tandem Differential Mobility Analysis (IV-TDMA) . J. Aerosol Sci. , 40 : 1019 – 1029 .
- Saleh , R. , Shihadeh , A. and Khlystov , A. 2010 . Effect of Aerosol Generation Method on Measured Saturation Pressure and Enthalpy of Vaporization of Dicarboxylic Acid Aerosols . Aerosol Sci. Technol. , 44 : 302 – 307 .
- Saleh , R. , Shihadeh , A. and Khlystov , A. 2011 . On Transport Phenomena and Evaporation Time Scales in Thermodenuders . Atmos. Meas. Tech. , 4 : 571 – 581 .
- Saleh , R. , Walker , J. and Khlystov , A. 2008 . Determination of Saturation Pressure and Enthalpy of Vaporization of Semi-Volatile Aerosols: The Integrated Volume Method . J. Aerosol Sci. , 39 : 876 – 887 .
- Saliba , N. , Moussa , S. , Salame , H. and El-Fadel , M. 2006 . Variation of Selected Air Quality Indicators Over the City of Beirut, Lebanon: Assessment of Emission Sources . Atmos. Environ. , 40 : 3263 – 3268 .
- Sioutas , C. , Kim , S. and Chang , M. 1999 . Development and Evaluation of a Prototype Ultrafine Particle Concentrator . J. Aerosol Sci. , 30 : 1001 – 1017 .
- Stanier , C. , Pathak , R. and Pandis , S. 2007 . Measurements and Volatility of Aerosols from a-pinene Ozonolysis . Environ. Sci. Technol. , 41 : 2756 – 2763 .
- Vaden , T. D. , Imre , D. , Beránek , J. , Shrivastava , M. and Zelenyuk , A. 2011 . Evaporation Kinetics and Phase of Laboratory and Ambient Secondary Organic Aerosol . Proc. Natl. Acad. Sci. USA , 108 ( 6 ) : 2190 – 2195 . doi: 10.1073/pnas.1013391108
- Virtanen , A. , Joutsensaari , J. , Koop , T. , Kannosto , J. , Yli-Pirila , P. , Leskinen , J. , Makela , J. M. , Holopainen , J. K. , Poschl , U. , Kulmala , M. , Worsnop , D. R. and Laaksonen , A. 2010 . An Amorphous Solid State of Biogenic Secondary Organic Aerosol Particles . Nature , 467 : 824 – 827 .
- Wehner , B. , Philippin , S. and Wiedensohler , A. 2002 . Design and Calibration of a Thermodenuder with an Improved Heating Unit to Measure the Size-Dependent Volatile Fraction of Aerosol Particles . J. Aerosol Sci. , 33 : 1087 – 1093 .
- Wilke , C. R. and Lee , C. Y. 1955 . Estimation of Diffusion Coefficients for Gases and Vapors . Ind. Eng. Chem. , 47 : 1253 – 1257 .