Abstract
Aqueous hydroxyl radical (∼10−12 M) oxidation of glycolaldehyde (1 mM), followed by droplet evaporation, forms secondary organic aerosol (SOA) that exhibits an effective liquid vapor pressure and enthalpy of vaporization of ∼10−7 atm and ∼70 kJ/mol, respectively, similar to the mix of organic acids identified in reaction samples. Salts of these acids have vapor pressures about three orders of magnitude lower (e.g., ammonium succinate ∼10−11 atm), suggesting that the gas–particle partitioning behavior of glycolaldehyde SOA depends strongly on whether products are present in the atmosphere as acids or salts. Several reaction samples were used to simulate cloud droplet evaporation using a vibrating orifice aerosol generator. Samples were also analyzed by ion chromatography (IC), electrospray ionization mass spectrometry (ESI-MS), IC-ESI-MS, and for total carbon. Glycolaldehyde SOA mass yields were 50–120%, somewhat higher than yields reported previously (40–60%). Possible reasons are discussed: (1) formation of oligomers from droplet evaporation, (2) inclusion of unquantified products formed by aqueous photooxidation, (3) differences in gas–particle partitioning, and (4) water retention in dried particles. These and similar results help to explain the enrichment of organic acids in particulate organic matter above clouds compared with those found below clouds, as observed previously in aircraft campaigns.
Copyright 2012 American Association for Aerosol Research
1. INTRODUCTION
Secondary organic aerosol (SOA) is a substantial contributor to organic particulate matter (PM) and is poorly captured by air quality models because its formation is not well understood (Turpin et al. Citation2000; EPA Citation2004; Heald et al. Citation2005; Kanakidou et al. Citation2005; Volkamer et al. Citation2006; Hallquist et al. Citation2009). Models that accurately link emissions to air pollution concentrations and effects are important to developing effective air quality management plans and understanding to what degree SOA is controllable (Carlton et al. Citation2010). SOA is formed from gas-phase chemistry, followed by either vapor pressure-based partitioning into particulate organic matter—SOAOM (Odum et al. Citation1996; Seinfeld and Pankow Citation2003; Hallquist et al. Citation2009) or aqueous-phase chemistry in clouds and wet aerosols—SOAaq (Blando and Turpin Citation2000; Gelencsér and Varga Citation2005; Ervens et al. Citation2011). Several article report comparable amounts of SOAOM and SOAaq globally and in certain regions, although uncertainties are large (Chen et al. Citation2007; Carlton et al. Citation2008; Fu et al. Citation2008, Citation2009; Gong et al. Citation2011). Furthermore, model results from Myriokefalitakis et al. (Citation2011) suggest that the vast majority of oxalate globally is formed through aqueous chemistry, making oxalate a good tracer for SOAaq. The enrichment of oxalate and organic acids above versus below cloud (Sorooshian et al. Citation2007a) and when aerosol liquid water content is high (Sorooshian et al. Citation2007b) provide atmospheric evidence for SOAaq.
SOAaq can form in clouds, fogs, and aerosol water; this work is focused on in-cloud formation. Briefly, during cloud processing, water-soluble organic gases dissolve into cloud water, undergo volume phase reactions, and form low-volatility compounds that remain in the particle phase when the droplets evaporate, thus forming SOAaq (Blando and Turpin Citation2000; Ervens et al. Citation2004, Citation2008, Citation2011; Lim et al. Citation2005; Heald et al. Citation2006; Loeffler et al. Citation2006; Sorooshian et al. Citation2006; El Haddad et al. Citation2009; De Haan et al. Citation2009a, Citation2009b). For example, the aqueous-phase hydroxyl (OH) radical oxidation (photooxidation) of glycolaldehyde, glyoxal, methylglyoxal, pyruvate, acetate, acetone, methacrolein, and methyl vinyl ketone directly or indirectly forms dicarboxylic acids and higher-molecular-weight compounds (HMWCs) (e.g., oligomers) (Altieri et al. Citation2006, Citation2008; Carlton et al. Citation2006, Citation2007; El Haddad et al. Citation2009; Liu et al. Citation2009; Michaud et al. Citation2009; Perri et al. Citation2009; Tan et al. Citation2009, Citation2010, Citation2011; Poulain et al. Citation2010; Zhang et al. Citation2010), with diacids forming preferentially in dilute solution (clouds) and HMWCs in concentrated solution (wet aerosols) (Tan et al. Citation2009; Ervens and Volkamer Citation2010; Lim et al. Citation2010). Several aqueous-phase photooxidation products (i.e., oxalate, pyruvate, glycolate, and HMWCs) are expected to stay, at least partially, in the particle phase once the cloud droplets evaporate and thus contribute to atmospheric organic PM.
While SOAaq formation from glyoxal and methylglyoxal has received more attention, glycolaldehyde is also a potentially important SOAaq precursor. Glycolaldehyde is produced in the gas phase from isoprene (∼6–22% molar yield; Lee et al. Citation2006; Nguyen et al. Citation2011), ethylene (20–100% molar yield; Niki et al. Citation1981; Orlando et al. Citation1998; Fu et al. Citation2008), methyl vinyl ketone (51–70% molar yields; Spaulding et al. Citation2003; Fu et al. Citation2008), and methylbutenol (50–78% molar yield; Atkinson and Arey Citation2003; Spaulding et al. Citation2003; Carrasco et al. Citation2007), and is directly emitted from biomass burning (4–20 Tg a−1; Yokelson et al. Citation1997; Akagi et al. Citation2011; Burling et al. Citation2011; Yokelson R. J., personal communication) and biofuel use (1–2 Tg a−1; Fu et al. Citation2008). Like the other SOAaq precursors, glycolaldehyde is a water-soluble compound (H* 298 > 3 × 105 M atm−1; Betterton and Hoffmann Citation1988). In the aqueous phase, it hydrates and reacts with OH radical to produce glycolic, glyoxylic, and oxalic acid, as well as glyoxal and HMWCs (Warneck Citation2003; Perri et al. Citation2009). The formation of malonic and succinic acid has also been reported (Perri et al. Citation2009). Perri et al. (Citation2009) estimated the SOA mass yield for glycolaldehyde by measuring products of the aqueous OH radical (∼10−13 M) oxidation of glycolaldehyde (1 mM), multiplying by the approximate fraction of each compound found in the particle phase in the atmosphere and dividing by the mass of precursor reacted. The total SOA yield was taken to be the sum of the individual compound yields. SOA mass yields were up to 60% for reaction times less than 25 min (e.g., cloud contact times) and about 40% at later times when glycolaldehyde was depleted (>40 min). The limitations of the approach used by Perri et al. (Citation2009) are that 14–23% of organic carbon was unquantified and therefore not included in the yield calculations, and that this approach neglects chemical transformations, if any, that occur during droplet evaporation. For example, glyoxal partially dehydrates during droplet evaporation and forms oligomers that could lead to higher SOA mass yields (Loeffler et al. Citation2006; De Haan et al. Citation2009b). Our current understanding of the processes that occur during cloud droplet evaporation is still incomplete and contributes to the uncertainty in SOA formation from cloud processing (Gong et al. Citation2011).
To our knowledge, El Haddad et al. (Citation2009) were the first to combine aqueous photooxidation and droplet evaporation. They reacted methacrolein with OH radicals, then nebulized and dried the sample solution in a mixing chamber. The major limitations of that study were: the need for substantial particle loss corrections, and that SOA yields were measured from samples after 5 h of reaction, whereas the lifetime of a cloud droplet is on the order of several minutes (Desboeufs et al. Citation2003; Ervens and Volkamer Citation2010).
The objectives of this article are to study the formation of glycolaldehyde SOA through cloud water chemistry (e.g., aqueous photooxidation) and droplet evaporation and to further the understanding of the gas–particle partitioning behavior of aqueous glycolaldehyde oxidation products. To accomplish this, we report experimental glycolaldehyde SOAaq yields at 10–13% relative humidity (RH) and compare the partitioning behavior of glycolaldehyde SOAaq with that of a suite of organic acids at a range of liquid vapor pressures (p L) and enthalpies of vaporization (ΔH vap). This article builds on work conducted by Perri et al. (Citation2009) and verifies that SOA forms from cloud processing of glycolaldehyde. The experimental approach used negates the need to correct for particle losses. To our knowledge, this is the first study to provide data characterizing the volatility of glycolaldehyde SOAaq.
2. EXPERIMENTAL METHODS
Detailed experimental procedures are provided below. Briefly, monodisperse droplets were generated from aqueous reaction solutions formed through the OH radical oxidation of glycolaldehyde and from standard solutions (). These droplets were evaporated and the diameters of the residual monodisperse aerosols were measured. The SOA yield was calculated as the mass of a residual particle divided by the mass of glycolaldehyde reacted from a single corresponding droplet. The ratio of the residual particle mass to the organic mass in the original droplet (PM mass/OM mass) is related to the fraction of the organic matter that remains in the particle phase. By comparing the PM mass/OC mass for reaction samples and for standards, the volatility of glycolaldehyde SOA was characterized. New insights into the aqueous-phase chemistry of glycolaldehyde are also provided.
FIG. 1 Experimental setup for aqueous photooxidation and droplet evaporation. Reaction samples were also analyzed by ion chromatography (IC), electrospray ionization mass spectrometry (ESI-MS), IC ESI-MS, and for total organic carbon (TOC). (Color figure available online.)
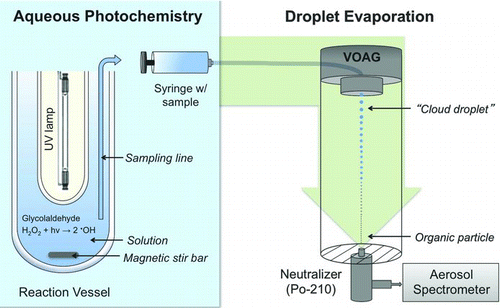
2.1. Aqueous-Phase Photochemistry
Aqueous photooxidation experiments were conducted with glycolaldehyde (1 mM) and OH radicals (∼10−12 M) in a 1-L reaction vessel, as described previously (Perri et al. Citation2009). Glycolaldehyde (98%; Pfaltz & Bauer) was dissolved with 18 Mohm Milli-Q water and diluted to 1 mM. OH radicals were formed in situ by photolysis of 5 mM hydrogen peroxide (diluted from 30% w/w; Sigma-Aldrich) using a 254-nm mercury lamp. Initial, final, and average OH radical concentrations were estimated to be 6 × 10−13 M, 4 × 10−12 M, and (4 ± 2) × 10−12 M, respectively, by modeling the chemistry in the reaction vessel using the mechanism published in Perri et al. (Citation2009) and (Citation2010). Experiments were conducted at 22 ± 3°C (n = 3). Reaction samples were collected at several reaction times from 0 to 118 min with 50–100% duplicates. The pH of the reaction solution decreased from 4.7 to 3.6 over the experiment, which is within typical cloud pH values (pH = 2–7; Pruppacher and Klett Citation1997; Warneck Citation2000). Samples were analyzed by ion chromatography (IC; Dionex ICS-3000) within 12 h of collection to quantify organic acids, as described previously (Perri et al. Citation2009; Tan et al. Citation2009). Samples were analyzed for total organic carbon (TOC; Shimadzu TOC-5000A), by electrospray ionization mass spectrometry (ESI-MS; HP-Agilent 1100), and by IC-ESI-MS, as described previously (Altieri et al. Citation2006; Perri et al. Citation2009; Tan et al. Citation2010).
While Perri et al. (Citation2009) added catalase to samples to destroy any remaining H2O2, we did not, so as to simplify the droplet evaporation experiments. Previous control experiments (Perri et al. Citation2009; Tan et al. Citation2009, Citation2010) have shown that glyoxylic acid degrades in the presence of H2O2, producing formic acid, while glycolic, oxalic, malonic, and succinic acid and glycolaldehyde do not. The reaction of glyoxylic acid with H2O2 is slow compared with its reaction with OH radicals and thus is not expected to affect the chemistry in the reaction vessel (Tan et al. Citation2009). However, this reaction converts glyoxylic to formic acid in samples awaiting analysis. Glycolaldehyde photolysis generates glycolic and glyoxylic acid; however, this reaction is also slow relative to OH radical oxidation (Perri et al. Citation2009). Thus, based on these past studies, we are confident that the experiments reported herein yield products generated from the reaction between glycolaldehyde and OH radicals, with the exception that the resulting samples are enriched in formic acid and depleted in glyoxylic acid.
2.2. Sample Solutions
In this study, droplet evaporation experiments (Section 2.3) were conducted using two types of solutions. These were used to validate previously estimated SOA yields, provide insights into the effects of cloud droplet evaporation, and characterize the volatility of the SOAaq formed. Solutions were: (1) samples from glycolaldehyde photooxidation experiments and (2) organic standards (individual and mixtures) that span a wide range of vapor pressures. Both types of solutions were used to generate monodisperse droplets that were then evaporated; the diameters of the residual particles were measured.
Reaction samples from the OH radical oxidation of glycolaldehyde were taken from the reaction vessel at specific reaction times (e.g., 0, 10, 20, 40, 50, and 70 min) using 25-mL syringes and passed through the droplet generation and evaporation system within 2–6 h (). Individual solutions of ammonium oxalate (99.0%; Fluka Analytical), oxalic (0.0991 N; Fluka Analytical), acetic (99.99%; Sigma-Aldrich), succinic (99.99%; Sigma-Aldrich), glutaric (99.9%; Aldrich Chemical), and tartaric (99.4%; Aldrich Chemical) acid were diluted to 0–4000 μM C.
2.3. Droplet Generation and Evaporation
A vibrating orifice aerosol generator (VOAG, TSI Model 3450; Berglund and Liu Citation1973) was used to generate and evaporate monodisperse droplets of sample solutions to form a monodisperse aerosol () (liquid flow rate 0.077 mL/min, frequency ∼160 kHz, dilution air 50 L/min, dispersion air 1000 mL/min, residence time 6 s, RH = 10–13%, T = ∼24°C). A major advantage of this approach is that one single-size droplet generates one single-size particle, facilitating the calculation of SOA mass yields by dividing the mass of one particle by the mass of precursor that reacted from one droplet. This approach is a relatively simple way of providing SOA mass yields in advance of the time when the aqueous and droplet evaporation chemistry is fully elucidated. The VOAG droplet generator was inverted and mounted on top of a vertical column to minimize coagulation. The VOAG passed filtered solutions (0.4 μm IsoporeTM membrane filter) through a 10-μm diameter (nominal) vibrating orifice, which produces a 20-μm nominal droplet diameter. Due to manufacturing tolerances, the orifice and droplet diameters may differ from the nominal values by ±25%; hence, the droplet diameter was determined by calibrating the system with ammonium sulfate, (NH4)2SO4 (3.1801 M; Fluka Analytical). The slope of D p versus C 1/3, from the relation D p = D d C 1/3, indicated the diameter of the generated droplets (18.3 ± 0.4 μm)—where D d is droplet diameter (μm), D p is particle diameter (μm), and C is the volumetric concentration of the solute in the solution (cm3 solute/cm3 solution). Succinic acid standards were used as an independent accuracy check. Deflection tests were performed routinely, deflecting the droplet stream with a perpendicular airstream to verify that the generated droplets were monodisperse (i.e., droplets remained in a single stream). Droplets were merged with a larger volume of clean dry air (Filtered Air Supply, TSI Model 3074B, two coalescing filters, membrane dryer, and carbon-vapor filter), which evaporated water and other volatile components of the droplets, leaving low-volatility particles (i.e., SOA). The particle residence time at 10–13% RH (6 s) is longer than typically used to equilibrate ambient particles in tandem differential mobility analyzer measurements of aerosol hygroscopicity, and is considered to be long enough for water equilibration, assuming an accommodation coefficient of 0.02 (Chan and Chan Citation2005); the potential for water retention is discussed below. Residual particles passed through a charge neutralizer (NRD, 2U500, Po-210) and their optical diameters were measured with an optical particle counter (OPC, Grimm Aerosol Spectrometer, Model 1.109, 31 channels) for 10 min after obtaining stable liquid feed pressure. To avoid organic contamination, the solutions, sheath/dilution air, and aerosol were transported through this system using Teflon tubing, with the exception of a short piece of flexible Tygon tubing (13 cm long, 0.3 cm wide) used to connect the OPC.
By knowing the precursor concentration, droplet diameter, resulting particle diameter, and approximate material density, we calculated the SOA mass yield, which is the mass of a residual particle (PM mass formed) divided by the mass of glycolaldehyde (GLYDE) reacted from the volume of solution contained in one droplet, in the following way:
TABLE 1 Particle geometric diameter (D p), concentration-weighted densities (ρ), SOAaq mass yields, and their corresponding uncertainty (Δ) as a function of reaction time (n ≥ 3, 1 mM glycolaldehyde and ∼10−12 M OH radicals)
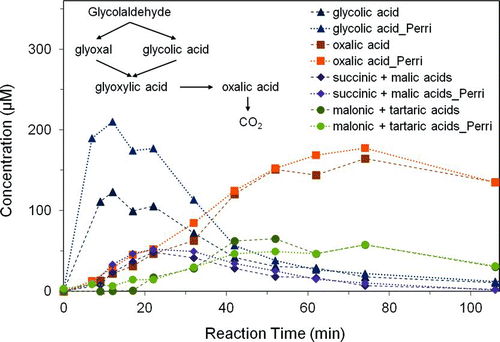
FIG. 2 Product concentrations from 1 mM glycolaldehyde and OH radicals (∼10−12 M), by ion chromatography (n = 3) for this work and for concentrations obtained by Perri et al. (Citation2009). Note that succinic plus malic acid as well as malonic plus tartaric acid co-elude and were quantified as succinic and malonic acid, respectively (Tan et al. Citation2009). Glyoxylic acid converts to formic acid in samples awaiting analysis (not shown). (Color figure available online.)
To assess the volatility of glycolaldehyde SOAaq, six dilutions of five organic standards and five dilutions of 10- and 40-min reaction solutions were sampled through the VOAG system (25 mL each). The TOC content of each solution ([TOC]droplet) and final particle diameter (D p) were directly measured, and from these, the mass of organic matter (OM) in the droplet and the mass of residual PM were calculated, respectively. [TOC]droplet was converted to OM mass(droplet) using OM/OC values (Table S1). D p was converted to PM mass by assuming spherical particles and using liquid densities (Table S1). Ratios of PM mass/droplet OM mass for reaction solutions and standards, and the liquid vapor pressures (p L°) and enthalpies of vaporization (ΔH vap) of the standards were used to characterize the volatility behavior of glycolaldehyde SOAaq. Note that ratios of PM mass/droplet OM mass represent the fraction of total droplet organic matter that remains in the particle phase (i.e., particle fraction), not to be confused with the SOA mass yields, which are defined differently. Values of p L° and ΔH vap for the standards were estimated using the SIMPOL group contribution method (Pankow and Asher Citation2008) and the Joback and Reid group contribution method (Joback and Reid Citation1987), respectively. Our experimental conditions were constant, stable, and controlled, so the differences between experiments are driven only by vapor pressure and hygroscopicity.
2.4. Quality Assurance and Quality Control
Measured organic acid concentrations were accurate within 6–10%, expressed as a pooled coefficient of variation based on independent standards, with the exception of formic (27%) and glyoxylic acid (60%). Calibration curves of conductivity (μS) versus concentration (M) had coefficients of determination (r2 ) better than 99.76% for all measured acids (glycolic, formic, glyoxylic, succinic, malonic, and oxalic acid). Method precision was 2%, expressed as a pooled coefficient of variation of samples (n = 240) collected in duplicate during experiments. Organic acid detection limits (μM) were 0.1–4.3 (Perri et al. Citation2009). Method precision (3%) for TOC (four injections/sample) was calculated as the pooled coefficient of variation of duplicate samples (n = 14). Variability of TOC measurements for identical time points across experiments was 14%.
Dynamic blanks were generated before each experiment by sampling Milli-Q water directly from the reaction vessel. Dynamic blanks were analyzed for organic acids and TOC and used to generate and evaporate droplets as if they were samples. IC analysis of dynamic blanks and water blanks confirmed no contamination. TOC values of dynamic blanks (1–35 μM C) were subtracted from their corresponding TOC sample measurements. The volume of the contaminants measured from the VOAG system was also subtracted from the samples using their corresponding dynamic blanks. The volume of contaminants from blanks was ∼0.01 μm3, about 6–27% of dried residual particle volume.
The performance of the VOAG was tested daily before and after each droplet evaporation experiment with (NH4)2SO4 standards. The performance criterion for acceptance was a ≤10% difference between the theoretical and the measured particle diameter. The method precision for diameter was 4%, calculated as the pooled standard deviation of the (NH4)2SO4 particle diameter divided by the mean diameter of 250 μM (NH4)2SO4 samples (n = 34) collected in duplicate during experiments. A 4% precision for particle diameter was also calculated based on 60 μM succinic acid solutions (n = 6).
3. RESULTS AND DISCUSSION
3.1. Glycolaldehyde Aqueous Photooxidation
Time profiles of product concentrations () and TOC (Figure S1) are in reasonable agreement with Perri et al. (Citation2009) and verify that compounds found predominantly in the particle phase in the atmosphere (e.g., oxalate, glycolate, malonate) can form from the aqueous photooxidation of glycolaldehyde with OH radicals. The only significant differences (p = 0.05, Cochran's t-test, two-tailed) between this work and Perri et al. (Citation2009) are that the concentrations of formic acid obtained in this work are higher, and those of glycolic and glyoxylic acid are lower, especially early in the reaction (∼12 min). The lower glyoxylic acid concentrations can be explained by the fact that we did not use catalase to destroy H2O2 in samples and glyoxylic acid reacts with H2O2 in samples awaiting analysis to form formic acid. This does not explain the lower glycolic acid concentrations, as recovery of glycolic acid was 93% more than 7 h after 250 μM H2O2 was added to a 250 μM glycolic acid standard (this work), in agreement with previous findings (Perri et al. Citation2009; Tan et al. Citation2009). Glycolic acid measurements reported herein are in good agreement with modeled glycolic acid (better than previous measurements; see in Perri et al. [Citation2009]). However, the difference between glycolic acid measurements is not well understood; these differences are a source of uncertainty in SOA mass yields for reaction times <30 min.
FIG. 3 (a) IC-ESI-MS spectra of a mixed standard: (A) glycolic acid (m/z − 75), (B) formic acid (not detectable by ESI-MS) and residual glycolic acid from peak A, (C) succinic acid (m/z − 117), (D) malonic acid (m/z − 103), (E) contaminants, including sulfate (m/z − 97), and (F) oxalic acid (m/z − 89). IC-ESI-MS spectra of samples taken from the reaction of 1 mM glycolaldehyde + OH radical at 17 min (b) and 52 min (c) reaction times: (A) glycolic acid (m/z − 75), (B) peak with retention time of formic acid, (C) succinic acid (m/z − 117) and malic acid (m/z − 133), (D) malonic acid (m/z − 103) and tartaric acid (m/z − 149), (E) peak with retention time of sulfate (m/z − 97), (F) oxalic acid (m/z − 89), and (G) high-molecular-weight compounds.
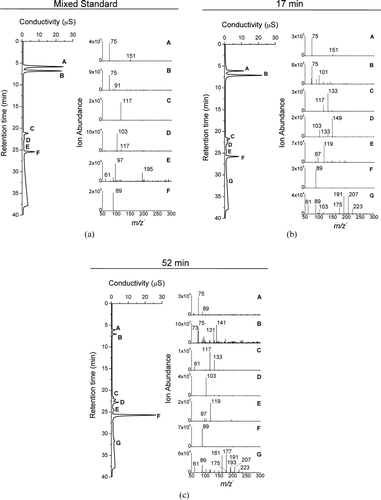
FIG. 4 (a) IC time profile of succinic and malic acid concentration from the reaction of 1 mM glycolaldehyde + OH radicals (∼10−12 M), and overlaid IC-ESI-MS ion abundance time profiles for m/z− 117 (succinic acid) and m/z− 133 (malic acid). (b) IC time profile of malonic and tartaric acid concentration, and overlaid IC-ESI-MS ion abundance time profiles for m/z− 103 (malonic acid) and m/z − 149 (tartaric acid). (Error bars represent the pooled coefficient of variation between experiments.)
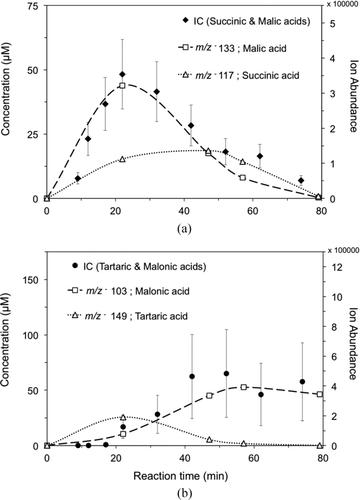
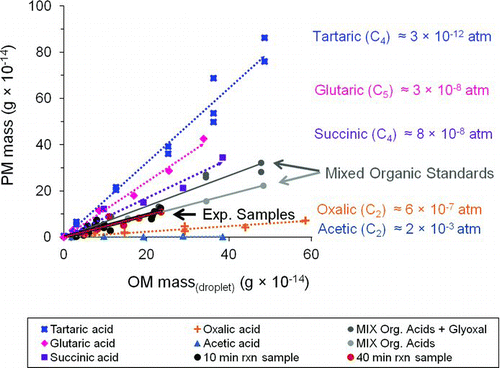
FIG. 5 Mass of residual particles (PM mass) formed from droplet evaporation of organic acid standard solutions of acetic, oxalic, succinic, glutaric, and tartaric acid. OM mass(droplet) is the mass of organic matter in the droplet. Labels include liquid vapor pressures estimated using the SIMPOL group contribution method (Pankow and Asher Citation2008). Slopes and r2 values are reported in (D d = 18.3 ± 0.4 μm; RH = 13 ± 2%; T = 24.1 ± 0.3°C). (Color figure available online.)
Experiments conducted with 1 mM glycolaldehyde provide insights into the OH radical oxidation of glycolaldehyde in clouds (1–5 μM glycolaldehyde) and in wet aerosols, where the total concentration of dissolved organics is quite high (1–10 M). Previous OH radical experiments conducted with 30, 300, and 3000 μM glyoxal (Tan et al. Citation2009) and the subsequent detailed chemical modeling from 10 μM to 10 M (Lim et al. Citation2010) suggest that organic radical–radical chemistry leading to higher-carbon-number products is minor at concentrations found in cloud water and becomes dominant at the high concentrations of water-soluble organics found in wet aerosols. We expect that this is also true for glycolaldehyde. The dilute glycolaldehyde chemistry model runs for the 1–5 μM glycolaldehyde (Perri et al. Citation2009) and the 1 mM glycolaldehyde experiments both show that the vast majority of the mass at the beginning of the reaction (10–20 min) is in the form of glycolic acid, glyoxylic (or formic) acid, and glyoxal (not measured here but quantified by Perri et al. [Citation2009]). Of these products, glycolic acid (and its salts) has the lowest volatility. Oxalic acid is the most abundant product after 30 min. Therefore, we expect that for typical cloud contact times (10–30 min), glycolaldehyde SOAaq formed through cloud processing will be predominantly glycolate and whatever HMWCs that form during droplet evaporation (e.g., glyoxal acetal oligomers; Loeffler et al. Citation2006; De Haan et al. 2009b).
Samples from experiments conducted with 1 mM glycolaldehyde also contained smaller concentrations of higher-molecular-weight products (IC-ESI-MS, ; ESI-MS online Supplemental Information, Figure S2) and products with higher carbon number than glycolaldehyde (). Since these products were formed in the presence and not in the absence of OH radicals, we expect that they formed through organic radical–radical chemistry, and that these and similar products will be the dominant products of glycolaldehyde chemistry in wet aerosols. It should be noted that while cloud contact times are typically 10–30 min, chemistry in aerosol water can proceed for hours with continuous addition of the precursor. IC-ESI-MS analyses ( and ) provide new insights into such chemistry.
The IC-ESI-MS negative-mode spectrum of a mixed standard solution and of experimental samples taken 17 and 52 min into the glycolaldehyde plus OH radical experiment are shown in . The mixed standard () consisted of glycolic acid (peak A, 5.8 min, m/z− 75), formic acid (peak B, 6.8 min, not detectable by ESI-MS), succinic acid (peak C, 21.1 min, m/z− 117), malonic acid (peak D, 22.3 min, m/z− 103), and oxalic acid (peak F, 25.4 min, m/z− 89). Contaminants, including sulfate (m/z− 97), can be found in peak E. Some glycolic acid can be seen in peak B. IC-ESI-MS results for experimental samples ( and c) verify the formation of glycolic and oxalic acid in the mechanism published in Lim et al. (Citation2005) and the formation of succinic and malonic acid published in Perri et al. (Citation2009) by IC alone. Interestingly, IC-ESI-MS shows that peaks with retention times of succinic and malonic acid also contain malic (peak C, m/z− 133) and tartaric acid (peak D, m/z− 149, 17 min sample only). HMWCs also form (peak G). Identification of HMWCs after IC separation verifies that it is not an artifact of electrospray ionization. In fact, most of the mass with retention time of succinic and malic acid is malic acid (m/z − 133). Malic acid peaks ∼20 min into the reaction, whereas succinic acid peaks after ∼50 min (). IC-ESI-MS () suggests that malonic acid (m/z− 103) is responsible for most of the mass with the retention time of “malonic and tartaric acid”; some tartaric acid (m/z− 149) is present early in the reaction (∼20 min). Tartaric acid formation has been observed also in the aqueous OH radical oxidation of glyoxal (3 mM), a glycolaldehyde intermediate (Tan et al. Citation2009). Its formation and concentration dynamics can be explained by organic radical–radical reactions (Tan et al. Citation2009; Lim et al. Citation2010). We expect that the formation of products with higher carbon numbers than glycolaldehyde (C2) (e.g., malic acid: C4, succinic acid: C4, tartaric acid: C4, and malonic acid: C3, and HMWCs), which are formed from glycolaldehyde in the presence and not the absence of OH radicals, are also formed through organic radical–radical reactions. Time profiles of other ions measured by IC-ESI-MS are provided in the online Supplemental Information (Figure S3).
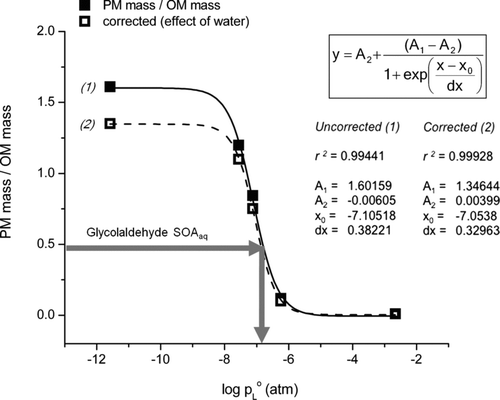
FIG. 6 Ratio of residual particle mass (PM mass) to droplet organic matter (OM mass) versus log(p L°), where p L° is the liquid vapor pressure. (PM mass/OM mass) values are the slopes from (see also ). (1) PM mass/OM mass values from , where densities were calculated from organic species. (2) PM mass/OM mass values computed using densities calculated with an upper-bound estimate of retained water. Gray arrow indicates the PM mass/OM mass of glycolaldehyde SOAaq.
3.2. Droplet Evaporation Experiments
3.2.1. Vapor Pressure and Enthalpy of Vaporization
The residual particle mass (PM mass) was well correlated with the mass of organic matter in the droplet (r2 = 0.84–0.99, and ) for all organic acid standards except acetic acid, which is quite volatile. We can observe from that as we go from the most volatile compound (acetic acid) to the least volatile compound (tartaric acid), the slope (m = PM mass/droplet OM mass) increases, indicating that a larger fraction of the mass remains in the particle phase. Shown also in are results of droplet evaporation experiments with 10- and 40-min reaction samples (thick black line: black and red in color figure online).
FIG. 7 Volume (D p 3) of residual particles and total organic carbon (TOC) in droplets, from droplet evaporation of oxalic acid and ammonium oxalate standard solutions. Liquid vapor pressure of ammonium oxalate p L° (US EPA: EPI Suite™ 2010) (D d = 18.3 ± 0.4 μm; RH = 13 ± 2%; T = 23.8 ± 0.3°C). The clear difference between oxalic acid and ammonium oxalate (and day-to-day reproducibiliy of oxalic and succinic acid results) provide confidence that the VOAG system was free of ammonium contamination.
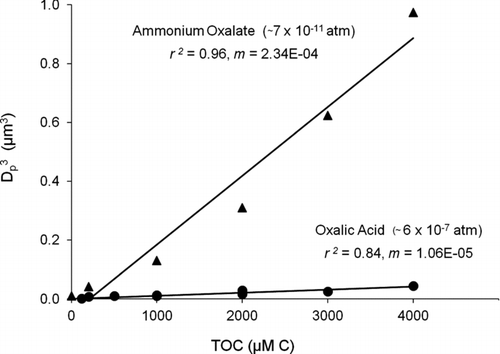
TABLE 2 Slopes (m), coefficients of determination (r2 ), liquid vapor pressures (p L°), and enthalpies of vaporization (ΔH vap)
The slopes (PM mass/droplet OM mass) from are plotted versus vapor pressure (p L°) in and versus enthalpy of vaporization (ΔH vap) in Figure S4 (). A sigmoidal curve was fit to these data in accordance with the gas–particle partitioning theory (Odum et al. Citation1996), since the slope (PM mass/droplet OM mass) reflects the fraction of OM found in the particle phase. These plots suggest that glycolaldehyde SOAaq behaves like a dicarboxylic acid, with a p L° of ∼10−7 atm and ΔH vap of ∼70 kJ/mol, and similar to the behavior of the mix of organic acids that comprise the majority of products identified in the reaction samples (). To our knowledge, this is the first study to characterize the volatility behavior of glycolaldehyde SOAaq.
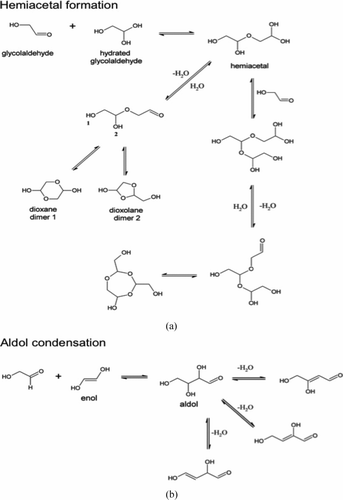
FIG. 8 Proposed mechanism for the formation of glycolaldehyde oligomers through hemiacetal formation (a) and aldol condensation (b).
Note that the ratio of the PM mass to droplet OM mass for tartaric acid is greater than 1 (, left-most data point). This is not surprising, since tartaric acid is expected to remain almost entirely in the particle phase, and it retains water even at low RH (5%; Peng et al. Citation2001). After accounting for the effect of water on particle density, suggests that the tartaric acid particles were <33% water. To understand to what degree water retension could alter , we present PM mass/OM mass for all standards calculated using dry densities (line 1, ) and densities assuming 33% water (line 2, ). This is an upper bound for the water fraction, since the other standards are not likely to retain as much water as tartaric acid. These corrections had a negligible effect on our characterization of the volatility behavior of glycolaldehyde SOAaq ().
We speculate that the vapor pressure of glycolaldehyde SOAaq will be orders of magnitude lower if the products are neutralized. For example, the liquid vapor pressure of succinic acid using the SIMPOL group contribution method (Pankow and Asher Citation2008) is 7.59 × 10−8 atm and the vapor pressure of ammonium succinate is 2 × 10−11 atm (EPA-EPI SuiteTM; EPA Citation2010). Enhancements in SOA yields in the presence of ammonia have been shown previously for α-pinene and ozone under dry (RH < 2%) and humid (RH = 50%) conditions (Na et al. Citation2007). Also, the work of Dinar et al. (Citation2008) verified that the reactive uptake of ammonia by acidic functional groups (e.g., adipic and citric acid) leads to the formation of ammonium salts and can substantially influence the chemical and physical properties of the aerosol. Interestingly, oxalate is found mostly in the particle phase in the atmosphere (Limbeck et al. Citation2001) even though the vapor pressure of oxalic acid is not that low. We suggest that this is because oxalate is mostly present in the atmosphere as a salt (e.g, ammonium oxalate) (). Certainly, the form of these acids depends on their pK a, the availability of ammonia and the abundance of stronger particle-phase acids (i.e., acidic sulfate), which impacts aerosol acidity. Oxalic acid is the strongest organic acid detected in glycolaldehyde SOAaq (pK a(1) = 1.23 and pK a(2) = 4.19), followed by malonic, tartaric, glyoxylic, malic, glycolic, and succinic acid. At pH 4.5, a typical value in cloud water, most of these acids are expected to be in their dissociated form, pH > pK a (e.g., glycolic, glyoxylic, tartaric, and oxalic acid) or amphiprotic form, pK a(1) < pH < pK a(2) (e.g., malic and succinic acid). Aerosol pH is not well characterized and depends not only on the concentrations of major inorganic and organic ions but also on the water content, buffering capacity, and gas–particle partitioning of many semivolatile compounds (Kerminen et al. Citation2001; Xue et al. Citation2011). Cation to anion ratios close to 1, suggesting a neutral aerosol, are frequently observed in the western United States and polluted rural and urban European areas (Kerminen et al. Citation2001). In contrast, there are days in Hong Kong when aerosol pH is less than 1, suggesting that even oxalate is present as an acid (Xue et al. Citation2011).
3.2.2. Aldehyde Oligomerization
In aqueous solutions, glyoxal species (i.e., hydrated monomers and oligomers) coexist in equilibrium and the predominant form depends on the concentration. Monomers dominate when glyoxal concentrations are below 1 M; at higher concentrations, dimers and oligomers dominate (Whipple Citation1970). Glyoxal reacts with itself to form acetal oligomers in evaporating droplets, thus contributing to SOA formation through cloud droplet evaporation (Loeffler et al. Citation2006; De Haan et al. Citation2009b). Similarly, during droplet evaporation, we expect the dehydration of glycolaldehyde to initiate the formation of glycolaldehyde oligomers via hemiacetal formation. We propose the formation of a hemiacetal that either forms dioxane and dioxolane dimers or reacts with another glycolaldehyde molecule to form an open-chain trimer, which in the process of drying, ultimately forms a trimer ring through intermolecular nucleophilic reactions (Baldwin Citation1976) (). Additionally, glycolaldehyde could oligomerize through aldol condensation (). We expect that glycolaldehyde oligomers formed in this way also contribute to the formation of SOA in the atmosphere.
3.2.3. SOAaq Mass Yields
The mass of SOAaq per mass of glycolaldehyde reacted (SOAaq mass yield) (, ) decreased gradually with time from about 120% to 50%. We expect that these early yields are driven by glycolic acid and oligomers formed during dehydration of glycolaldehyde and glyoxal. Later yields are driven by oxalic acid (). Shown in are the yields from this work (squares), yields estimated by Perri et al. (triangles), and the yields that would have been obtained if all droplet organic matter had remained in the particle phase (circles). These were calculated using IC quantification of organic acids and model predictions of remaining glycolaldehyde and glyoxal. Note that these values (circles) underestimate the true upper bound at later reaction times, since for later reaction times (>40 min), unquantified products accounted for about 14–23% of TOC in the reaction vessel (Perri et al. Citation2009). The fact that yields for early time points are much lower than would be obtained if all droplet organic matter were retained in the particle phase and yields for later time points are not, is consistent with the fact that the solution contained more volatile components (formic acid, glycolaldehyde, glyoxal) for early time points compared with later time points. This finding also suggests that glycolaldehyde and glyoxal are not 100% retained in the particle phase through oligomer formation.
FIG. 9 SOAaq mass yields from the reaction of 1 mM glycolaldehyde + OH radicals (∼10−12 M). Squares are mass yields calculated in this study using concentration-weighted densities and assuming spherical particles (n ≥ 3; D d = 18.3 ± 0.4 μm; RH = 10 ± 1%; T = 23.7 ± 0.7°C). Triangles are yields estimated by Perri et al. (Citation2009) using concentrations of species measured in the reaction vessel and estimating the fraction of each that would remain in the particle phase from atmospheric measurements. Circles are upper-bound yields obtained if all the organic mass in the droplet remained in the residual particle, calculated using IC quantification of organic acids and model predictions of remaining glycolaldehyde and glyoxal and neglecting unquantified products. (Error bars for open squares represent the pooled coefficient of variation for identical time points across experiments. Error bars for circles are from error propagation).
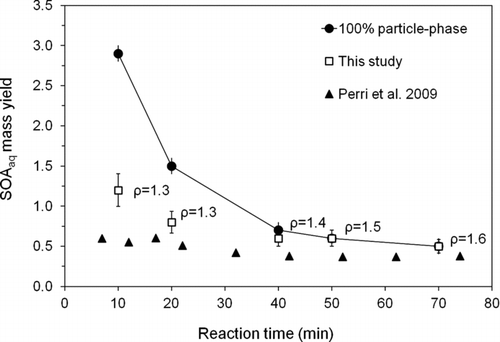
Our yields are higher than SOAaq mass yields estimated by Perri et al. (Citation2009). There are several possible explanations for this. First, Perri's yields were calculated based only on species quantified by IC, whereas the yields in this work also included any unquantified products from the photooxidation reaction and droplet evaporation (e.g., acetal oligomers). Second, Perri's yields were calculated assuming no water retention, whereas our yields include any particle-bound water. Such water might exist in equilibrium with its vapor, or particles could exist in a metastable state after drying, with a kinetic barrier inhibiting the release of water, despite the theoretically adequate time for water equilibration.
Differences between yields obtained herein and those in Perri et al. (Citation2009) could also occur because of differences in gas–particle partitioning or effects of residual H2O2 in samples. Perri et al. made use of atmospheric measurements of gas–particle partitioning to estimate the fraction of each product in the particle phase, whereas in this work, the gas–particle partitioning was determined by experimental conditions and might not be the same as in the atmosphere. Also, this work did not use catalase and hence residual H2O2 could have formed complexes with glyoxal and converted glyoxylic acid to formic acid. Specifically, Lee et al. (Citation2011) found that about 15–20% of glyoxal is consumed by H2O2 within 2 h. Tan et al. (Citation2010) found that H2O2 converted 98% of glyoxylic acid to the more volatile formic acid within 2 h. Last, the OPC was calibrated by the manufacturer with polystyrene latex (PSL) particles, which have a refractive index of 1.59. Based on measured products, we expect that glycolaldehyde SOA has a refractive index of ∼1.5, introducing an uncertainty of ∼10% in the measured particle diameter, before accounting for the effects of retained water on refractive index.
4. CONCLUSIONS
Mass yields were measured for SOAaq formed from the aqueous OH radical (∼10−12 M) oxidation of glycolaldehyde (1 mM) and the volatility behavior was characterized. While SOAaq mass yields are expected to vary with atmospheric conditions, vapor pressure and enthalpy of vaporization can be used to evaluate the behavior of this material under a range of atmospheric conditions (e.g., temperature dependence). This work verifies that SOAaq forms after glycolaldehyde reacts with OH radicals in droplets. Additional chemistry during droplet evaporation enhances the SOAaq production (e.g., oligomerization of aldehydes). SOAaq yields were highest (∼80–120%) at reaction times (∼10–20 min) that are most relevant to cloud droplet lifetimes. These yields include the contribution of HMWCs and account for droplet evaporation. Glycolaldehyde SOAaq behaves like a dicarboxylic acid, with a liquid vapor pressure of ∼10−7 atm and the enthalpy of vaporization of ∼70 kJ/mol. However, we expect the vapor pressure to be considerably lower if the mix of organic acids in the SOAaq gets neutralized in the atmosphere to form organic salts.
uast_a_686676_sup_26079883.zip
Download Zip (948.4 KB)Acknowledgments
This research was supported, in part, by the Ford Foundation Dissertation Fellowship Award sponsored by the Ford Foundation and administered by the National Research Council of the National Academies, the APERG (Air Pollution Educational and Research Grant) program administered by the MASS-A&WMA (Mid-Atlantic States Section of the Air and Waste Management Association), the GAANN (Graduate Assistance in Areas of National Need) Project P200A060156 Interdisciplinary Graduate Education in Environmental Science and Engineering, NSF-ATM-0630298 grant, NOAA (grant NA07OAR4310279), and EPA-STAR (Environmental Protection Agency-Science To Achieve Results) (RD-83375101–0). The authors thank Dr Neil Donahue, Ron Lauck, and Gabriel J. Reyes-Rodríguez for their contributions.
[Supplementary materials are available for this article. Go to the publisher's online edition of Aerosol Science and Technology to view the free supplementary files.]
Related Research Data
REFERENCES
- Akagi , S. K. , Yokelson , R. J. , Wiedinmyer , C. , Alvarado , M. J. , Reid , J. S. Karl , T. 2011 . Emission Factors for Open and Domestic Biomass Burning for Use in Atmospheric Models . Atmos. Chem. Phys. , 11 : 4039 – 4072 .
- Altieri , K. E. , Carlton , A. G. , Lim , H. J. , Turpin , B. J. and Seitzinger , S. P. 2006 . Evidence for Oligomer Formation in Clouds: Reactions of Isoprene Oxidation Products . Environ. Sci. Technol. , 40 : 4956 – 4960 .
- Altieri , K. E. , Seitzinger , S. P. , Carlton , A. G. , Turpin , B. J. , Klein , G. C. and Marshall , A. G. 2008 . Oligomers Formed through In-Cloud Methylglyoxal Reactions: Chemical Composition, Properties, and Mechanisms Investigated by Ultra-High Resolution FT-ICR Mass Spectrometry . Atmos. Environ. , 42 : 1476 – 1490 .
- Atkinson , R. and Arey , J. 2003 . Gas-Phase Tropospheric Chemistry of Biogenic Volatile Organic Compounds: A Review . Atmos. Environ. , 37 ( Suppl. 2 ) : S197 – S219 .
- Baldwin , J. E. 1976 . Rules for Ring Closure . J. Chem. Soc. Chem. Commun. , 18 : 734 – 736 .
- Berglund , R. N. and Liu , B. Y. H. 1973 . Generation of Monodisperse Aerosol Standards . Environ. Sci. Technol. , 7 : 147 – 153 .
- Betterton , E. A. and Hoffmann , M. R. 1988 . Henry Law Constants of Some Environmentally Important Aldehydes . Environ. Sci. Technol. , 22 : 1415 – 1418 .
- Blando , J. D. and Turpin , B. J. 2000 . Secondary Organic Aerosol Formation in Cloud and Fog Droplets: A Literature Evaluation of Plausibility . Atmos. Environ. , 34 : 1623 – 1632 .
- Burling , I. R. , Yokelson , R. J. , Akagi , S. K. , Urbanski , S. P. , Wold , C. E. Griffith , D. W. T. 2011 . Airborne and Ground-Based Measurements of the Trace Gases and Particles Emitted by Prescribed Fires in the United States . Atmos. Chem. Phys. , 11 : 12197 – 12216 .
- Carlton , A. G. , Pinder , R. W. , Bhave , P. V. and Pouliot , G. A. 2010 . To What Extent Can Biogenic SOA Be Controlled? . Environ. Sci. Technol. , 44 : 3376 – 3380 .
- Carlton , A. G. , Turpin , B. J. , Altieri , K. E. , Seitzinger , S. P. , Mathur , R. Roselle , S. J. 2008 . CMAQ Model Performance Enhanced When In-Cloud SOA Is Included: Comparisons of OC Predictions with Measurements . Environ. Sci. Technol. , 42 : 8798 – 8802 .
- Carlton , A. G. , Turpin , B. J. , Altieri , K. E. , Seitzinger , S. , Reff , A. Lim , H.-J. 2007 . Atmospheric Oxalic Acid and SOA Production from Glyoxal: Results of Aqueous Photooxidation Experiments . Atmos. Environ. , 41 : 7588 – 7602 .
- Carlton , A. G. , Turpin , B. J. , Lim , H. J. , Altieri , K. E. and Seitzinger , S. 2006 . Link between Isoprene and Secondary Organic Aerosol (SOA): Pyruvic Acid Oxidation Yields Low Volatility Organic Acids in Clouds . Geophys. Res. Lett. , 33 : L06822
- Carrasco , N. , Doussin , J. F. , O’Connor , M. , Wenger , J. C. , Picquet-Varrault , B. Durand-Jolibois , R. 2007 . Simulation Chamber Studies of the Atmosphere Oxidation of 2-Methyl-3-Buten-2-ol: Reaction with Hydroxyl Radicals and Ozone under a Variety of Conditions . J. Atmos. Chem. , 56 : 33 – 55 .
- Chan , M. N. and Chan , C. K. 2005 . Mass Transfer Effects in Hygroscopic Measurements of Aerosol Particles . Atmos. Chem. Phys. , 5 : 2703 – 2712 .
- Chen , J. , Griffin , R. J. , Grini , A. and Tulet , P. 2007 . Modeling Secondary Organic Aerosol Formation through Cloud Processing of Organic Compounds . Atmos. Chem. Phys. , 7 : 5343 – 5355 .
- De Haan , D. O. , Corrigan , A. L. , Smith , K. W. , Stroik , D. R. , Turley , J. J. Lee , F. E. 2009a . Secondary Organic Aerosol-Forming Reactions of Glyoxal with Amino Acids . Environ. Sci. Technol. , 43 : 2818 – 2824 .
- De Haan , D. O. , Corrigan , A. L. , Tolbert , M. A. , Jimenez , J. L. , Wood , S. E. and Turley , J. J. 2009b . Secondary Organic Aerosol Formation by Self-Reactions of Methylglyoxal and Glyoxal in Evaporating Droplets . Environ. Sci. Technol. , 43 : 8184 – 8190 .
- Desboeufs , K. V. , Losno , R. and Colin , J. L. 2003 . Relationship between Droplet pH and Aerosol Dissolution Kinetics: Effect of Incorporated Aerosol Particles on Droplet pH during Cloud Processing . J. Atmos. Chem. , 46 : 159 – 172 .
- Dinar , E. , Anttila , T. and Rudich , Y. 2008 . CCN Activity and Hygroscopic Growth of Organic Aerosols following Reactive Uptake of Ammonia . Environ. Sci. Technol. , 42 : 793 – 799 .
- El Haddad , I. , Yao , L. , Nieto-Gligorovski , L. , Michaud , V. , Temime-Roussel , B. Quivet , E. 2009 . In-Cloud Processes of Methacrolein under Simulated Conditions – Part 2: Formation of Secondary Organic Aerosol . Atmos. Chem. Phys. , 9 : 5107 – 5117 .
- EPA . 2004 . Air Quality Criteria for Particulate Matter , Environmental Protection Agency (EPA), Research Triangle Park, NC .
- EPA . 2010 . Estimation Programs Interface Suite™ for Microsoft® Windows, v 4.00 , Washington , DC : Environmental Protection Agency (EPA) .
- Ervens , B. , Carlton , A. G. , Turpin , B. J. , Altieri , K. E. , Kreidensweis , S. M. and Feingold , G. 2008 . Secondary Organic Aerosol Yields from Cloud-Processing of Isoprene Oxidation Products . Geophys. Res. Lett. , 35 : L02816
- Ervens , B. , Feingold , G. , Frost , G. J. and Kreidenweis , S. M. 2004 . A Modeling Study of Aqueous Production of Dicarboxylic Acids: 1. Chemical Pathways and Speciated Organic Mass Production . J. Geophys. Res. , 109 : D15205
- Ervens , B. , Turpin , B. J. and Weber , R. J. 2011 . Secondary Organic Aerosol Formation in Cloud Droplets and Aqueous Particles (aqSOA): A Review of Laboratory, Field and Model Studies . Atmos. Chem. Phys. , 11 : 11069 – 11102 .
- Ervens , B. and Volkamer , R. 2010 . Glyoxal Processing by Aerosol Multiphase Chemistry: Towards a Kinetic Modeling Framework of Secondary Organic Aerosol Formation in Aqueous Particles . Atmos. Chem. Phys. , 10 : 8219 – 8244 .
- Fu , T. M. , Jacob , D. J. and Heald , C. L. 2009 . Aqueous-Phase Reactive Uptake of Dicarboyls as a Source of Organic Aerosol over Eastern North America . Atmos. Environ. , 43 : 1814 – 1822 .
- Fu , T. M. , Jacob , D. J. , Wittrock , F. , Burrows , J. P. , Vrekoussis , M. and Henze , D. K. 2008 . Global Budgets of Atmospheric Glyoxal and Methylglyoxal, and Implications for Formation of Secondary Organic Aerosols . J. Geophys. Res.-Atmos. , 113 : D15303
- Gelencsér , A. and Varga , Z. 2005 . Evaluation of the Atmospheric Significance of Multiphase Reactions in Atmospheric Secondary Organic Aerosol Formation . Atmos. Chem. Phys. , 5 : 2823 – 2831 .
- Gong , W. , Stroud , C. and Zhang , L. 2011 . Cloud Processing of Gases and Aerosols in Air Quality Modeling . Atmosphere , 2 : 567 – 616 .
- Hallquist , M. , Wenger , J. C. , Baltensperger , U. , Rudich , Y. , Simpson , D. Claeys , M. 2009 . The Formation, Properties and Impact of Secondary Organic Aerosol: Current and Emerging Issues . Atmos. Chem. Phys. , 9 : 5155 – 5236 .
- Heald , C. L. , Jacob , D. J. , Park , R. J. , Russell , L. M. , Huebert , B. J. Seinfeld , J. H. 2005 . A Large Organic Aerosol Source in the Free Troposphere Missing from Current Models . Geophys. Res. Lett. , 32 : L18809
- Heald , C. L. , Jacob , D. J. , Turquety , S. , Hudman , R. C. , Weber , R. J. Sullivan , A. P. 2006 . Concentrations and Sources of Organic Carbon Aerosols in the Free Troposphere over North America . J. Geophys. Res. , 111 : D23S47
- Joback , K. G. and Reid , R. C. 1987 . Estimation of Pure-Component Properties from Group-Contributions . Chem. Eng. Commun. , 57 : 233 – 243 .
- Kanakidou , M. , Seinfeld , J. H. , Pandis , S. N. , Barnes , I. , Dentener , F. J. Facchini , M. C. 2005 . Organic Aerosol and Global Climate Modelling: A Review . Atmos. Chem. Phys. , 5 : 1053 – 1123 .
- Kerminen , V.-M. , Hillamo , R. , Teinilä , K. , Pakkanen , T. , Allegrini , I. and Sparapani , R. 2001 . Ion Balances of Size-Resolved Tropospheric Aerosol Samples: Implications for the Acidity and Atmospheric Processing of Aerosols . Atmos. Environ. , 35 : 5255 – 5265 .
- Lee , A. , Goldstein , A. H. , Kroll , J. H. , Ng , N. L. , Varutbangkul , V. Flagan , R. C. 2006 . Gas-Phase Products and Secondary Aerosol Yields from the Photooxidation of 16 Different Terpenes . J. Geophys. Res.-Atmos. , 111 : D17305
- Lee , A. K. Y. , Zhao , R. , Gao , S. S. and Abbatt , J. P. D. 2011 . Aqueous-Phase OH Oxidation of Glyoxal: Application of a Novel Analytical Approach Employing Aerosol Mass Spectrometry and Complementary Off-Line Techniques . J. Phys. Chem. A. , 115 : 10517 – 10536 .
- Lim , H. J. , Carlton , A. G. and Turpin , B. J. 2005 . Isoprene Forms Secondary Organic Aerosol through Cloud Processing: Model Simulations . Environ. Sci. Technol. , 39 : 4441 – 4446 .
- Lim , Y. B. , Tan , Y. , Perri , M. J. , Seitzinger , S. P. and Turpin , B. J. 2010 . Aqueous Chemistry and Its Role in Secondary Organic Aerosol (SOA) Formation . Atmos. Chem. Phys. , 10 : 10521 – 10539 .
- Limbeck , A. , Puxbaum , H. , Otter , L. and Scholes , M. C. 2001 . Semivolatile Behavior of Dicarboxylic Acids and Other Polar Organic Species at a Rural Background Site (Nylsvley, RSA) . Atmos. Environ. , 35 : 1853 – 1862 .
- Liu , Y. , El Haddad , I. , Scarfogliero , M. , Nieto-Gligorovski , L. , Temime-Roussel , B. Quivet , E. 2009 . In-Cloud Processes of Methacrolein under Simulated Conditions – Part 1: Aqueous Phase Photooxidation . Atmos. Chem. Phys. , 9 : 5093 – 5105 .
- Loeffler , K. W. , Koehler , C. A. , Paul , N. M. and de Haan , D. O. 2006 . Oligomer Formation in Evaporating Aqueous Glyoxal and Methyl Glyoxal Solutions . Environ. Sci. Technol. , 40 : 6318 – 6323 .
- Michaud , V. , El Haddad , I. , Yao , Liu , Sellegri , K. , Laj , P. Villani , P. 2009 . In-Cloud Processes of Methacrolein under Simulated Conditions – Part 3: Hygroscopic and Volatility Properties of the Formed Secondary Organic Aerosol . Atmos. Chem. Phys. , 9 : 5119 – 5130 .
- Myriokefalitakis , S. , Tsigaridis , K. , Mihalopoulos , N. , Sciare , J. , Nenes , A. Kawamura , K. 2011 . In-Cloud Oxalate Formation in the Global Troposphere: A 3-D Modeling Study . Atmos. Chem. Phys. , 11 : 5761 – 5782 .
- Na , K. , Song , C. , Switzer , C. and Cocker , D. R. 2007 . Effect of Ammonia on Secondary Organic Aerosol Formation from Alpha-pinene Ozonolysis in Dry and Humid Conditions . Environ. Sci. Technol. , 41 : 6096 – 6102 .
- Nguyen , T. B. , Roach , P. J. , Laskin , J. , Laskin , A. and Nizkorodov , S. A. 2011 . Effect of Humidity on the Composition of Isoprene Photooxidation Secondary Organic Aerosol . Atmos. Chem. Phys. , 11 : 6931 – 6944 .
- Niki , H. , Maker , P. D. , Savage , C. M. and Breitenbach , L. P. 1981 . An FTIR Study of Mechanisms for the HO Radical Initiated Oxidation of C2H4 in the Presence of NO: Detection of Glycolaldehyde . Chem. Phys. Lett. , 80 : 499 – 503 .
- Odum , J. R. , Hoffmann , T. , Bowman , F. , Collins , D. , Flagan , R. C. and Seinfeld , J. H. 1996 . Gas/Particle Partitioning and Secondary Organic Aerosol Yields . Environ. Sci. Technol. , 30 : 2580 – 2585 .
- Orlando , J. J. , Tyndall , G. S. , Bilde , M. , Ferronato , C. , Wallington , T. J. Vereecken , L. 1998 . Laboratory and Theoretical Study of the Oxy Radicals in the OH- and Cl-Initiated Oxidation of Ethene . J. Phys. Chem. , 102 : 8116 – 8123 .
- Pankow , J. F. and Asher , W. E. 2008 . SIMPOL.1: A Simple Group Contribution Method for Predicting Vapor Pressures and Enthalpies of Vaporization of Multifunctional Organic Compounds . Atmos. Chem. Phys. , 8 : 2773 – 2796 .
- Peng , C. , Chan , M. N. and Chan , C. K. 2001 . The Hygroscopic Properties of Dicarboxylic and Multifunctional Acids: Measurements and UNIFAC Predictions . Environ. Sci. Technol. , 35 : 4495 – 4501 .
- Perri , M. J. , Lim , Y. B. , Seitzinger , S. P. and Turpin , B. J. 2010 . Organosulfates from Glycolaldehyde in Aqueous Aerosols and Clouds: Laboratory Studies . Atmos. Environ. , 44 : 2658 – 2664 .
- Perri , M. J. , Seitzinger , S. and Turpin , B. J. 2009 . Secondary Organic Aerosol Production from Aqueous Photooxidation of Glycolaldehyde: Laboratory Experiments . Atmos. Environ. , 43 : 1487 – 1497 .
- Poulain , L. , Katrib , Y. , Isikli , E. , Liu , Y. , Wortham , H. Mirabel , P. 2010 . In-Cloud Multiphase Behaviour of Acetone in the Troposphere: Gas Uptake, Henry's Law Equilibrium and Aqueous Phase Photooxidation . Chemosphere , 81 : 312 – 320 .
- Pruppacher , H. R and Klett , J. D. 1997 . Microphysics of Clouds and Precipitation () , 2nd ed. , Dordrecht , , The Netherlands : Kluwer Academic Publishers .
- Seinfeld , J. H. and Pankow , J. F. 2003 . Organic Atmospheric Particulate Material . Annu. Rev. Phys. Chem. , 54 : 121 – 140 .
- Sorooshian , A. , Brechtel , F. J. , Ervens , B. , Feingold , G. , Varutbangkul , V. Bahreini , R. 2006 . Oxalic Acid in Clear and Cloudy Atmospheres: Analysis of Data from International Consortium for Atmospheric Research on Transport and Transformation 2004 . J. Geophys. Res. , 111 : D23S45
- Sorooshian , A. , Lu , M.-L. , Brechtel , F. J. , Jonsson , H. , Feingold , G. Flagan , R. C. 2007a . On the Source of Organic Acid Aerosol Layers above Clouds . Environ. Sci. Technol. , 41 : 4647 – 4654 .
- Sorooshian , A. , Ng , N. L. , Chan , A. W. H. , Feingold , G. , Flagan , R. C. and Seinfeld , J. H. 2007b . Particulate Organic Acids and Overall Water-Soluble Aerosol Composition Measurements from the 2006 Gulf of Mexico Atmospheric Composition and Climate Study (GoMACCS) . J. Geophys. Res. , 112 : D13201
- Spaulding , R. S. , Schade , G. W. , Goldstein , A. H. and Charles , M. J. 2003 . Characterization of Secondary Atmospheric Photooxidation Products: Evidence for Biogenic and Anthropogenic Sources . J. Geophys. Res. , 108 D8, 4247
- Tan , Y. , Carlton , A. G. , Seitzinger , S. P. and Turpin , B. J. 2010 . SOA from Methylglyoxal in Clouds and Wet Aerosols: Measurement and Prediction of Key Products . Atmos. Environ. , 44 : 5218 – 5226 .
- Tan , Y. , Lim , Y. B. , Altieri , K. E. , Seitzinger , S. P. and Turpin , B. J. 2011 . Mechanisms Leading to Oligomers and SOA through Aqueous Photooxidation: Insights from OH Radical Oxidation of Acetic Acid and Methylglyoxal . Atmos. Chem. Phys. , 12 : 801 – 813 .
- Tan , Y. , Perri , M. J. , Seitzinger , S. P. and Turpin , B. J. 2009 . Effects of Precursor Concentration and Acidic Sulfate in Aqueous Glyoxal-OH Radical Oxidation and Implications for Secondary Organic Aerosol . Environ. Sci. Technol. , 43 : 8105 – 8112 .
- Turpin , B. J. and Lim , H. J. 2001 . Species Contributions to PM2.5 Mass Concentrations: Revisiting Common Assumptions for Estimating Organic Mass . Aerosol Sci. Technol. , 35 : 602 – 610 .
- Turpin , B. J. , Saxena , P. and Andrews , E. 2000 . Measuring and Simulating Particulate Organics in the Atmosphere: Problems and Prospects . Atmos. Environ. , 34 : 2983 – 3013 .
- Volkamer , R. , Jimenez , J. L. , San Martini , F. , Dzepina , K. , Zhang , Q. Salcedo , D. 2006 . Secondary Organic Aerosol Formation from Anthropogenic Air Pollution: Rapid and Higher than Expected . Geophys. Res. Lett. , 33 : L17811
- Warneck , P. 2000 . Chemistry of the Natural Atmosphere () , 2nd ed. , San Diego , CA : Academic Press .
- Warneck , P. 2003 . In-Cloud Chemistry Opens Pathway to the Formation of Oxalic Acid in the Marine Atmosphere . Atmos. Environ. , 37 : 2423 – 2427 .
- Whipple , E. B. 1970 . Structure of Glyoxal in Water . J. Am. Chem. Soc. , 90 ( 24 ) : 7183 – 7186 .
- Xue , J. , Lau , A. K. H. and Yu , J. Z. 2011 . A Study of Acidity on PM2.5 in Hong Kong using Online Ionic Chemical Composition Measurements . Atmos. Environ. , 45 : 7081 – 7088 .
- Yokelson , R. J. , Susott , R. , Ward , D. E. , Reardon , J. and Griffith , D. W. T. 1997 . Emissions from Smoldering Combustion of Biomass measured by Open-Path Fourier Transform Infrared Spectroscopy . J. Geophys. Res. , 102 : 18865 – 18877 .
- Zhang , X. , Chen , Z. M. and Zhao , Y. 2010 . Laboratory Simulation for the Aqueous OH-Oxidation of Methyl Vinyl Ketone and Methacrolein: Significance to the In-Cloud SOA Production . Atmos. Chem. Phys. , 10 : 9551 – 9561 .