
Abstract
Differential mobility analyzers (DMAs) are widely used for calibrating other instruments and measuring aerosol size distributions. DMAs classify aerosol particles according to their electrical mobility, which is assumed to be constant during the classification process. However, particles containing semivolatile substances can change their size in the DMA, leading to sizing errors. In this article, the effect of particle size changes during the classification process on the sizing accuracy of DMAs is discussed. It is shown that DMAs select particles whose time-of-flight-averaged electrical mobility is equal to that of stable particles that are selected under given operating conditions. For evaporating particles, this implies that DMAs select particles that are originally larger than the reported size. At the exit of the DMA, selected particles are smaller than the reported size. Particle evaporation and growth inside DMAs was modeled to study the effect of particle size changes on the sizing accuracy and the transfer function of DMAs in constant- and scanning-voltage modes of operation. Modeling predictions were found to agree well with the results of experiments with ammonium nitrate aerosol. The model was used to estimate sizing errors when measuring hygroscopic and other volatile aerosols. Errors were found to be larger at smaller sizes and low sheath flow rates. Errors, however, are fairly small when saturation concentration is below 10 μg/m3, assuming an evaporation coefficient of 0.1. Particles size changes during classification lead to distortion of the DMA transfer function. In voltage scanning mode, errors are generally larger, especially at high scan rates.
Copyright 2014 American Association for Aerosol Research
1 INTRODUCTION
Differential mobility analyzers (DMAs), also called electrostatic classifiers, are widely used in experimental aerosol science to measure aerosol size distributions (Fissan et al. Citation1983; ten Brink et al. Citation1983; Wang and Flagan Citation1990), aerosol hygroscopic growth (Swietlicki et al. Citation2008), kinetics of particle evaporation and condensation (Rader and McMurry Citation1986), and to produce monodisperse aerosols for calibrating other instruments (Knutson Citation1976).
DMAs classify aerosol particles according to their mobility in an electric field (Knutson and Whitby Citation1975). The most common type of DMA consists of two concentric metal cylinders (Knutson and Whitby Citation1975; Winkelmayr et al. Citation1991), between which particle-free sheath air flows. Sampled air is introduced as a narrow band along the sheath flow and near the outer cylinder of the DMA. By applying a voltage difference between the outer (grounded) and the inner cylinders, an electrical field is created that causes charged particles to migrate through the sheath air toward the inner cylinder. A part of the sheath flow is extracted through a narrow slit at the end of the inner cylinder. Only particles in a narrow mobility range, which for particles of the same charge implies a very narrow size range, leave the classifier in the extracted stream.
A well-defined relationship exists between the size of transmitted particles, their charge, and the electrical field in the DMA (Knutson and Whitby Citation1975; Wang and Flagan Citation1990). This relationship assumes that particles do not change their size during passage through the classifier. However, if there are differences in temperature, RH, and/or gaseous concentrations between sampled air and conditions in the DMA, particles containing volatile or hygroscopic compounds can evaporate or grow inside the DMA. Differences in the gas concentrations can also be induced by losses of volatile species, such as nitric acid, to the walls of the DMA and the tubing used to supply sheath air to the DMA. Particle size changes during classification can lead to sizing errors. Since laboratory-generated and ambient aerosols often contain semivolatile and hygroscopic compounds (Robinson et al. Citation2007; Swietlicki et al. Citation2008), sizing error due to particle growth or shrinkage could significantly affect the accuracy of data collected using DMAs.
In this article, I describe a simple method for estimating sizing error when measuring volatile aerosols using DMAs. The method is tested against measurements of ammonium nitrate aerosol and a particle trajectory model that has also been developed in this study. The particle trajectory model is used to estimate sizing errors and their effect on the DMA transfer function in both constant- and scanning-voltage modes of operation. I discuss the sensitivity of errors to sheath air flow rate, voltage scan rate, and aerosol volatility. Two special cases, ammonium nitrate and hygroscopic aerosol measurements, are discussed in more detail. I also provide general guidelines for minimizing sizing errors when measuring volatile aerosols.
2 THEORY
A theoretical analysis is presented here to provide an insight into the effect of aerosol volatility on the sizing accuracy of DMAs. Several simplifying assumptions are employed in this analysis. Following the approach taken by Wang and Flagan (Citation1990), the flow field in the DMA is assumed to be radially uniform. Aerosol and monodisperse flows are assumed to be infinitely thin. Nonidealities of the flow and electric field at the entrance and exit regions of the DMA are neglected. Only particles of a single initial electrical mobility in a constant electrical field are considered. The effect of particle evaporation or growth on the shape of the DMA transfer function is not considered in this analysis because the transfer function is assumed to be infinitely thin, but is studied using a particle trajectory model that is discussed in a separate section below.
2.1 Classification of Stable Particles
The radial electric field in the DMA, as defined by Wang and Flagan (Citation1990), is:
[1] in which V is the voltage applied to the DMA, r is the radial position of the particle, and r1 and r2 are the radii of the inner and outer cylinder, respectively. E1 = V/(r1ln (r1/r2)) is the electric field at the surface of the inner cylinder.
The radial movement of a particle in an electric field can be described as:
[0002] in which t is time and Zp is the particle electrical mobility. The particle electrical mobility is defined as:
[3] in which dp is the particle diameter, i is the number of charges on the particle, e is the elementary charge, μ is the gas viscosity, and Cc(dp) is the Cunningham slip correction factor. The following expression for the Cunningham slip correction was used in this study (Allen and Raabe Citation1985):
[4] in which Kn = 2λa/dp is the Knudsen number, with λa being the mean free path of air molecules.
For a particle to exit through the slit in the central cylinder, i.e., be selected by the DMA, it must traverse the distance between the entrance point (rin) and the inner cylinder (r1) in exactly the time it takes to reach the exit, tf. Under the employed assumptions, this time is equal to the mean gas residence time:
[5] in which L is the length of the classification region, Qa and Qsh are the aerosol and sheath flow rates, respectively.
Integrating Equation (Equation2[0002] ) from rin to r1 provides an expression for the particle mobility that will be selected at the given time of flight and electrical field in the DMA:
[6] in which Zp, n is the mobility of a stable particle that is selected at the given conditions (the “nominal” mobility).
2.2 Classification of Volatile Particles
If a particle evaporates or grows in the DMA, its mobility becomes a function of time due to the changes of the particle size:
[7] The particle size change rate due to differences in vapor concentration between the particle surface and the sheath air are described by (Seinfeld and Pandis Citation2006):
[8] in which dp is the particle diameter, D is the diffusion coefficient of the evaporating species, Cs is the mole-fraction-averaged saturation vapor concentration of all volatile species in the particle, Csh is the total concentration of all the volatile species in the sheath air, ρ is the density of the particle, F(dp, α) is the Fuchs–Sutugin correction factor, K(dp, σ) is the Kelvin effect correction, α is the accommodation/evaporation coefficient, and σ is surface tension. Expressions for the Fuch-Sutugin and the Kelvin terms and an explanation of the use of mole-fraction-averaged and total vapor concentrations to describe the behavior of a mixture can be found in the online supplemental information (SI).
The radial movement of a particle in an electric field should also take into account the time-dependence of its electrical mobility:
[9]
Under the approximation of a uniform flow field and an infinitely thin aerosol flow, the criterion for classification remains the same as for stable particles: to be classified, particles need to reach the central rod in exactly the mean gas residence time, tf. By integrating Equation (Equation9[9] ), an expression for the particle mobility that will be selected at given operating conditions (i.e., time of flight and electrical field) can be obtained:
[10] in which
is the time-of-flight-averaged particle mobility.
It should be noted that the right-hand side of Equation (Equation10[10] ) is equal to the nominal mobility, i.e., the mobility of a stable particle that is selected under the given conditions, Zp, n:
[11] The above equation provides an important insight into DMA classification of volatile particles. For a volatile particle to be selected by the DMA, its average electrical mobility during the time it spends in the DMA classification region must equal that of a nonvolatile particle that is selected under the same conditions. The size of this stable, singly charged particle will be hereafter referred to as the “nominal diameter” and designated as dp, n.
Equation (Equation10[10] ) offers a fairly simple way of estimating sizing error when measuring volatile aerosol, if the time evolution of particle mobility in the DMA classification region can be modeled. As an example, particle evaporation in vapor-free sheath air is discussed in the following section.
2.3 Implications for Sizing Accuracy
To consider the implications of the theory for DMA sizing accuracy, let us consider an evaporating particle. Due to evaporation, the size of the particle decreases during its passage through the DMA classification region, leading to a corresponding increase in its electrical mobility (Equation (Equation7[7] )). Because of their increased mobility, particles of the nominal size at the entrance of the DMA will reach the central cylinder before the exit slit and thus will not be selected by the DMA. Therefore, in order to be classified, particles need to be initially larger than the nominal size, i.e., have lower initial mobility. This constraint implies DMAs that report nominal diameters will always underestimate the original size (i.e., diameter before entering the DMA) of evaporating particles. This is demonstrated in . To satisfy the criteria for classification given by Equation (Equation11
[11] ), the average mobility of evaporating particles needs to be equal to that of nominal size particles. Since the initial size of evaporating particles needs to be larger than the nominal size, this requires the transmitted (final) diameter to be smaller than the nominal size. Thus, if one uses the DMA for calibrating other instruments, the transmitted particles will be smaller than the expected (nominal) size. For growing particles the opposite will be true, i.e., the initial size will be smaller than the nominal size, while the transmitted size will be larger.
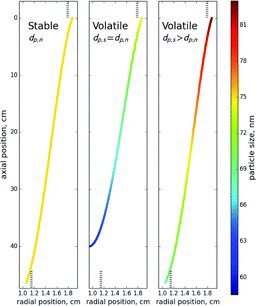
Let us consider evaporation in vapor-free sheath air to demonstrate how particle volatility affects sizing error. To simplify analysis, the effect of particle evaporation on vapor concentration in sheath air is neglected. Under these simplifications, Equation (Equation8[8] ) transforms into:
[12] Let us also assume that particles are composed of a single volatile component, such that Cs and ρ remain constant during evaporation. Given these assumptions, Equation (Equation12
[12] ) can be used to substitute variables in Equation (Equation10
[10] ):
[13] in which dp, s and dp, t are the particle diameter at the entrance (sampled diameter) and exit of the DMA (transferred diameter), respectively.
Recalling Equation (Equation11[11] ), the right hand side of Equation (Equation13
[13] ) is equal to the particle nominal mobility (Equation (Equation3
[3] )):
[14]
Simplifying Equation (Equation14[14] ), we obtain the relationship between nominal diameter, dp, n, and the initial size of the evaporating particle, dp, s:
[15]
Equation (Equation15[15] ) shows that the parameter ρ/(4DCstf) controls sizing error, not only because of its explicit presence in the equation, but also because it controls the final size of the particles, dp, t, as is apparent from Equation (Equation8
[8] ). Therefore, larger sizing errors are to be expected for lower sheath flow rates (larger tf values) and higher volatility compounds (larger Cs values).
3 MODELING
Two models have been developed to assess the effect of particle size changes during the classification process on the sizing accuracy of DMAs. One model (a “simplified model”) uses equations derived in Section 2 to estimate sizing error at a constant DMA voltage as a function of particle size and volatility. The second model (a “trajectory model”) was developed to investigate the effect of aerosol volatility on the sizing error and the shape of transfer function for both constant voltage and scanning modes of operation. Unlike the simplified model, the trajectory model accounts for fully developed laminar flow and finite-width aerosol and monodisperse flows to describe particle trajectories inside the DMA. The results of the trajectory model were also used to assess the performance of the simplified model. Both models are described in the following text.
3.1 Simplified Model
The simplified model was developed to estimate the sensitivity of DMA sizing accuracy to aerosol volatility and to compare the predictions of the theory with observations. The effect of aerosol evaporation or growth on vapor concentration in sheath air is neglected. In the case of particle evaporation, the vapor concentration in sheath air is assumed to be zero. It is unrealistic to study all possible internally mixed aerosol compositions that can be encountered in ambient and laboratory-generated aerosols. Therefore, only single-component aerosols were modeled. The volatility of compounds was chosen to be within the volatility range reported for ambient and laboratory aerosols, with Cs values ranging from 10− 2μg/m3 to 102μg/m3 (Donahue et al. Citation2006). All calculations presented here were made for the TSI long DMA, as it is one of the most widely used DMA types.
To calculate the relationship between dp, n and dp, s for different values of Cs, the following steps were taken. For a set of dp, s and a given Cs, a set of dp, t diameters was calculated by numerically integrating Equation (Equation12[12] ) from 0 to tf. The obtained set of (dp, s, dp, t) pairs was then used to calculate the integral in Equation (Equation15
[15] ) to find the corresponding values of dp, n.
For comparison with experiments that used ammonium nitrate (NH4NO3) aerosol (see Experimental section), the equilibrium constant of NH4NO3 was calculated for the temperature and relative humidity of the experiments. Following Dassios and Pandis (Citation1999), the equilibrium constant of NH4NO3 was calculated using the expression proposed by Mozurkewich (Citation1993) and assuming the particles to be in a metastable aqueous state. Cs was calculated as the square root of the equilibrium constant. The values of the diffusion coefficient D (0.11 cm2/s), surface tension σ (0.12 ergs/cm2), and evaporation coefficient α (0.6) were taken from Dassios and Pandis (Citation1999).
Since the experiments (see Section 4) involve two DMAs, the nominal diameter that would be reported by the second DMA was calculated in the following way. For each particle diameter entering the first DMA (dp, s), the final (dp, t), and the nominal (dp, n) diameters were obtained. The calculated dp, t values were used as initial diameters in calculating the nominal diameters for the second DMA. The ratio of the nominal diameters of the second to those of the first DMAs was calculated. The calculated ratios were then compared to ratios observed during experiments.
3.2 Particle Trajectory Model
A particle trajectory model was developed to evaluate the performance of the simplified model and to study the effect of particle volatility on the shape of the DMA transfer function. Similarly to the simplified model, aerosol was assumed to be composed of a single volatile compound and sheath air was assumed to be vapor free. It was also assumed that particle evaporation or growth does not affect vapor concentration in sheath air. However, unlike the simplified model, fully-developed laminar flow in the DMA was assumed. The effect of particle diffusion on particle trajectories inside the DMA was ignored. Diffusion effects are significant only for particles smaller than 10 nm (Kousaka et al. Citation1986; Stolzenburg Citation1988). As will be shown in Section 5, aerosol volatility has a significant effect at much larger sizes. All calculations were performed for the TSI long DMA with the sheath and excess flow rates fixed at 3 L/min, and aerosol and monodisperse flow rates fixed at 0.3 L/min. For the scanning voltage mode of operation only up-scans (exponentially increasing voltage) were considered. Down-scans (decreasing voltage) are rarely used in practice because of stronger transfer function distortion than in up-scans (Collins et al. Citation2004). Two scan durations (30 s and 300 s) for a full voltage scan (10 V to 10,000 V) were tested. These scan rates are representative of the fastest and slowest scans allowed by TSI Scanning Mobility Particle Sizer (SMPS) software.
The particle trajectory model follows the approach adopted by Hagwood et al. (Citation1999) and Collins et al. (Citation2004). Instead of modeling individual air streamlines, virtual slits for the entrance of aerosol flow and the exit of monodisperse flow are used. These virtual slits are depicted by dotted lines in . The radial position of virtual slits is chosen such that they define regions through which an appropriate portion of flow passes (0.3 L/min in these simulations).
For the constant-voltage mode of operation, the model solves the following set of ordinary differential equations:
[16]
[17]
[18] in which x is the axial coordinate and u(r) is the velocity at radial position r. The radial velocity distribution for the fully-developed laminar flow is given by (Bird et al. Citation1960):
[19] in which y = r/r2, k = r1/r2, and uav is the average gas velocity in the DMA.
For the scanning mode of operation, Equation (17) is modified to take into account changes in the electrical field during the voltage scan (Wang and Flagan Citation1990):
[20] in which E1, 0 = V0/(r1ln (r1/r2)) is the electric field at the surface of the inner cylinder at the start of the scan, τ = ts/log (V1/V0) is the scan time constant, ts is the scan time, and V0 and V1 are the voltages at the start and the end of the scan, respectively.
Particles of equal initial size and charge are introduced at x = 0 at 50 equally spaced radial positions between the entrance virtual slit and r2. The number of particles at each radial position ri is set to be proportional to the fraction of aerosol flow passing through this radial position, i.e., proportional to riu(ri). Particle position is then tracked through the DMA. Particles that at the end of the DMA (x = L) are radially between the exit virtual slit and r1 are marked as selected particles. This procedure is repeated for several particle sizes, and the concentration and initial and final sizes of each selected particle are recorded.
In conventional DMA analysis, the transfer function is defined as the probability of a particle of a given mobility to be selected and transferred through the DMA (Knutson and Whitby Citation1975). As discussed in Section 2, sizes of selected and transferred particles are not the same for a volatile aerosol. Therefore, two separate probability functions need to be defined—the “selection” function and the “transfer” function. The selection function, Ωs(dp), is the probability of selecting a particle whose size at the entrance of the DMA is dp. The transfer function, Ωt(dp), is the probability of finding a particle of size dp at the exit of the DMA. The selection function is calculated by binning the selected particles according to their initial size. Likewise, the transfer function is determined by binning the model results according to the final particle size.
Modeling of the scanning mode of operation is similar to the constant-voltage mode, but the procedure is repeated by introducing particles at 0.01 s intervals for the duration of the scan (ts). The counts, initial size and final size of particles that satisfy the selection criteria (the radial position at x = L is between the exit virtual slit and r1), are accumulated into 0.1-s-wide time bins. Time bin 0.1 s is the integration time for the counts of selected particles that is used in TSI SMPS software. Mixing effects in the tubing and the CPC are neglected in this analysis. For each time bin, the selection and transfer functions are calculated by binning counts of the selected particles according to their initial and final sizes, respectively.
4 EXPERIMENTAL
Modeling results were compared with experiments using laboratory-generated NH4NO3 aerosol. NH4NO3 is a semivolatile compound that dissociates into gaseous nitric acid and ammonia (Mozurkewich Citation1993). Nitric acid is a very reactive compound and can be easily stripped from the gas when transported through sample tubing (Neuman et al. Citation1999). This can happen both in the sheath lines and in the DMA itself leading to partial or complete removal of nitric acid from the sheath air. This will cause particle evaporation inside the DMA even if the temperature and RH are equilibrated with sample air.
A detailed description of the aerosol generation and dilution system used in this study can be found elsewhere (ten Brink et al. Citation2000). Ammonium sulfate ((NH4)2SO4) and NH4NO3 aerosols were used for the experiments. Because (NH4)2SO4 is a stable compound, it was used as a reference. The aerosols were produced by nebulizing a water solution of the corresponding salts with a Wiesbadener Doppelinhalator (Wiesbadener Inhalatoren-Vertrieb, Wiesbaden, Germany). The produced aerosol was mixed with an excess of particle-free, dry, pressurized air which resulted in an RH below 15%. The air was then led through a column of about 1 m3 internal volume filled with glass rings to insure complete mixing and drying of the aerosol. The aerosol stream was then introduced into a tunnel where it was diluted with 500 L/min of particle-free air (approximately 30% RH). The resulting number size distribution was centered at approximately 75 nm in diameter with the total number concentration around 10,000 cm− 3.
The sizing accuracy of classifiers was tested using a Tandem DMA (TDMA) setup. A diagram of the TDMA setup is shown in . The TDMA consisted of two identical electrostatic classifiers (TSI EC-3071, TSI “long DMA”) in series. The flow settings in both of the classifiers were identical. The sheath air flow rate was set to 3 L/min and was 10 times higher than the aerosol flow rate. At these flow settings, the residence time inside each DMA is 7.36 s. The sheath air of the classifiers was not recirculated and was taken from the room through a silica gel diffusion dryer to bring it to 30% RH. A HEPA filter was used to remove particles from sheath air.
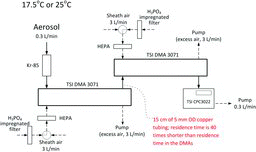
The monodisperse output of the first DMA was connected to the inlet of the second DMA using stainless steel tubing (15 cm long 0.5 cm OD). Connecting tubing was, like the rest of the TDMA, at room temperature. The residence time in the connecting tubing was approximately 40 times shorter than the combined residence time in the two classifiers. Consequently, evaporation of NH4NO3 during the transport through the connecting tubing can be neglected. Aerosol was sampled through a Kr-85 neutralizer (Model3077, TSI Inc.) into the first classifier. No neutralizer was used between the DMAs. The monodisperse output from the second classifier was directed to a condensation particle counter (CPC, Model CPC3022, TSI Inc.). The first classifier was maintained at a certain (constant) voltage, which changed from experiment to experiment to select different particle sizes. Aerosol exiting the first classifier was measured with the second DMA, which was operated in the scanning mode using AIM software (Version 2, TSI Inc.).
While the accuracy of the scanning mode can deviate from that of a constant-voltage operation, slow voltage scan rates provide accuracy close to that of constant voltage measurements (Collins et al. Citation2004). In this study, low scan rates were achieved by performing measurements within a narrow size range that covered the output of the first DMA with the longest scan duration allowed by the software (300 s per scan). The accuracy of the scanning mode depends on the ratio of the gas mean residence time in the DMA, tf, to the scan time constant, τ (Collins et al. Citation2004):
[21] In this study,
ranged from 0.0177 to 0.0567. At such slow scan rates, the scanning DMA transfer function is practically identical to that of the constant-voltage operation (Collins et al. Citation2004). Therefore, the second DMA could be assumed to operate in quasiconstant voltage mode. This allowed simplifying data analysis.
Raw data counts collected during scans were inverted using an author-written data inversion program based on the Twomey algorithm (Twomey Citation1977). Since all particles exiting the first DMA are charged and a neutralizer was not used between the DMAs, the inversion does not correct for particle charging efficiency. All particles were assumed to be singly charged. This assumption was used because the second DMA cannot distinguish singly from multiply charged stable particles since they have the same electrical mobility.
Ammonia is a dissociation product of NH4NO3. High concentrations of ammonia may slow down evaporation of ammonium nitrate. The concentration of gaseous ammonia was measured using AMANDA automated ammonia monitor (Wyers et al. Citation1993). The concentration of gaseous ammonia in these experiments was between 12 and 20 μg/m3. To test the effect of ammonia concentration in sheath air on experimental results, ammonia was removed from the sheath air using phosphoric acid impregnated filters in one set of experiments. The efficiency of the impregnated filters was tested using AMANDA. The ammonia concentration behind the filter was found to be less than 0.03 μg/m3. The ammonia concentration in sheath air is expected to be lower, because filters should be more efficient at lower flow rates than those used in the AMANDA (30 L/min). Ammonia was not removed from the sample air. Aerosol flow comprises 9% of total flow in the DMA. Thus, even if one assumes complete mixing of sheath air and sample air in the DMA, ammonia concentration in these experiments should be about 11 times lower than in sample air. Actual ammonia levels in sheath air in the classification region are likely to be lower because the two air streams are not mixed, meaning that ammonia can be introduced into sheath air only by molecular diffusion.
During most of the experiments the room air temperature was 25°C and was stable within 0.5°C. To test the influence of temperature via its effect on the equilibrium constant of ammonium nitrate, the instruments were also operated at 17.5°C.
To test the accuracy and stability of the DMAs, (NH4)2SO4 aerosol was measured before and after each experiment with ammonium nitrate aerosol. The measured size of (NH4)2SO4 aerosol was taken as the nominal particle size for both of the DMAs. The size distribution and mean size of measured (NH4)2SO4 aerosol transmitted through the first DMA was compared with that of ammonium nitrate aerosol. If evaporation of NH4NO3 within the DMAs affects their sizing accuracy, the measured NH4NO3 distribution would be expected to deviate from that of ammonium sulfate aerosol.
It should be noted that any differences between NH4NO3 and (NH4)2SO4 observed in this experimental setup are only due to particle evaporation that occurs in the two DMAs. Evaporation that occurs prior to the first DMA does not affect results as long as particles do not evaporate completely prior to being sampled by the first DMA. If a particle does not change its size inside the DMAs, both DMAs should give similar classification, i.e., the second DMA should report the size corresponding to the voltage of the first DMA, whether or not the particle has evaporated prior to sampling. Since the residence time in the connection between the two DMAs was approximately 40 times shorter than the combined residence time in the DMAs, evaporation in the connecting tubing can be neglected. Evaporation of particles after leaving the second DMA does not affect sizing either. Sizing is based only on the voltage at which particles exiting the second DMA are detected. Evaporation after the second classifier may affect the results only if the particles evaporate to such an extent that they cannot be detected by the CPC (smaller than about 7 nm in diameter for CPC3022), in which case no counts would be detected. Thus, in this setup, any discrepancies between measured sizes of volatile and stable aerosols are due to processes occurring inside the DMAs only, and are unaffected by evaporation outside the DMAs.
5 RESULTS AND DISCUSSION
5.1 Comparison of Simplified and Trajectory Models
The simplified model was compared to the trajectory model. The trajectory model uses a finite width aerosol flow and produces output in the form of sampled and reported (i.e., in terms of the nominal size that corresponds to the sampled size) size distributions, as described in Section 3.2. The simplified model uses an infinitely thin flow approximation and thus produces a single combination of sampled and reported sizes. To make the comparison between the two models possible, the output of the trajectory model was converted to mean particle sizes. The initial radial position of aerosol in the simplified model (rin in Equations (Equation6[6] ) and (Equation10
[10] )) was taken as the midpoint between the entrance virtual slit and r2. Ratios of nominal to sampled sizes were calculated and compared between the models. The trajectory model results for the constant voltage mode of operation were taken as a reference. The dependence of these ratios on particle size and volatility will be discussed in the following sections.
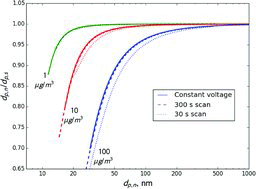
shows a comparison of simplified and trajectory model results. It is remarkable that the simplified model, which assumes a constant electrical field in the DMA, produced virtually identical results to those of the trajectory model for the constant voltage mode of operation. The results for a 300 s scan over a voltage range of 10 V–10,000 V () were also very close to those calculated for the constant voltage mode. This is expected, because the DMA performance at low values of
(long scan times) is similar to its performance in the constant voltage mode of operation (Collins et al. Citation2004). The scan rates used in experiments with NH4NO3 aerosol were even slower than analyzed in that study (
between 0.0177 and 0.0567, see Section 4). Therefore, the results obtained during those experiments could well be approximated as those from two DMAs operated under constant-voltage mode, simplifying interpretation of experimental results (see Section 5.2).
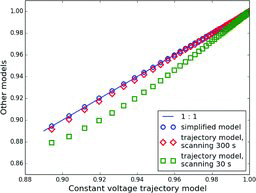
A faster scan (30 s, ) produces a significantly larger deviation from the constant-voltage mode of operation, with deviation increasing at smaller values of the ratio. This is because faster scans are more affected by the radial dependence of the flow velocity in the DMA (Collins et al. Citation2004). As will be shown in Section 5.3, deviation of the modeled ratios for 30-s scans from those calculated for the constant-voltage mode depends on particle size and aerosol volatility, with deviations being generally larger for smaller sizes and more volatile aerosols.
Overall, it can be concluded that the simplified model produces a very adequate approximation of dp, n for the constant voltage mode of operation and for slow (300 s over 10 V–10,000 V voltage range) voltage scans. Thus, Equation (Equation10[10] ) can be used to obtain a fairly accurate estimate of sizing error when measuring volatile particles. In the case of a single component aerosol whose growth or evaporation does not affect the vapor concentration in the sheath air, Equation (Equation15
[15] ) can be used.
5.2 Comparison with Observations
The simplified model predictions were compared to the results of TDMA experiments with NH4NO3 aerosol (see Section 4). presents a compilation of five experiments at 25°C in which the first DMA was set to five different voltages. It shows size distributions of ammonium sulfate and NH4NO3 aerosols transmitted by the first DMA as measured by the second DMA. The midpoint sizes measured at each voltage are summarized in . The size of ammonium sulfate particles measured at each voltage was taken to be the nominal size corresponding to that voltage.
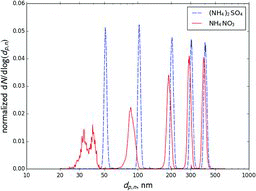
Table 1 Summary of measured midpoint sizes of ammonium sulfate (AS) and ammonium nitrate (AN) aerosol during different experiments. All sizes are in nm
It is clear that the mean size of transmitted NH4NO3 aerosol is measured consistently smaller than that of (NH4)2SO4 aerosol (the set nominal size). Deviation from nominal size increases and the classifier output broadens as nominal size decreases. Broadening of measured distributions could be explained by different evaporation rates at different particle sizes. The finite width of the DMA transfer function could contribute to this phenomenon, because smaller particles evaporate more relative to larger particles. The modeled effect of particle evaporation on the transfer function will be discussed in Section 5.6. However, part of the observed broadening could also be due to the presence of doubly charged particles. Because no neutralizer was used between the DMAs, the data were inverted under the assumption that all particles transmitted by the first DMA are singly charged. The presence of doubly charged particles becomes apparent in measurements of ammonium nitrate at 51.3 nm nominal size. Two peaks were observed, the larger one apparently corresponding to doubly charged particles that were transmitted by the first DMA, as will be discussed at the end of this section.
Results of the experiments, where ammonia was removed from sheath air, were practically identical to those when ammonia concentration in sheath air was between 12 and 20 μg/m3 ( and ). This indicates that nitric acid concentration in sheath air was virtually zero, such that the product of ammonia and nitric acid concentrations that controls the evaporation rate becomes insensitive to ammonia concentration.
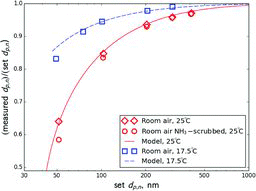
When measurements were performed at 17.5°C, the measured deviations from the nominal size were lower than those measured at 25°C ( and ). Due to the lower temperature, Cs of NH4NO3 is smaller, resulting in smaller deviations from the nominal size.
also shows the ratios of nominal diameters observed in the second DMA to those set by the first DMA modeled for the conditions of the experiments described above. Very good agreement with all of the experiments is observed. This provides strong support for the theory, allowing it to be used for estimating error per single DMA and extrapolating it to other compounds and operating conditions.
The simplified model also accurately predicts the size of the second peak observed during measurements of NH4NO3 at the set nominal size of 51.3 nm (). The size of stable doubly charged particles that corresponds to singly charged particles of 51.3 nm is 83.0 nm. The model predicts that these doubly charged particles will be detected by the second DMA as 40.3 nm singly charged particles. This agrees very well with the measured mean size of the second peak, which is 39.8 nm. Doubly charged particles at larger sizes are not well separated from singly charged particles and are, thus, not as apparent as at 51.3 nm nominal size. For example, at a set nominal size of 51.3 nm, singly charged particles appear in the second DMA to be 24.4% smaller than doubly charged particles. At a set nominal size of 103.2 nm, this difference is only 7.7%, which is smaller than the resolving power of the DMA (as defined by the width of the transfer function). The two peaks thus merge and appear as one broad peak in the measurements.
5.3 Predicted Error for Single DMA Measurements
It should be noted that the undersizing observed in the TDMA experiments is a combined result of evaporation in the two DMAs. Most measurements, such as size distribution measurements or monodisperse aerosol generation for instrument calibration, use a single DMA. In this section, the effect of aerosol volatility on the magnitude of sizing error in a single DMA is examined. Since there is a great multitude of semivolatile compounds, error will vary widely depending on compound volatility, surface tension, affinity of the vapor to the parts of the DMA, etc. It is impossible to analyze all possible scenarios. However, to give the reader a sense of the magnitude and importance of these errors, the following calculations were made.
To estimate the magnitude of error when measuring aerosol of different volatilities using a single DMA, the following is assumed. (1) Sheath air is assumed to be vapor-free. While this assumption provided good agreement with measurements of NH4NO3 aerosol, one should keep in mind that this assumption provides, most probably, an upper estimate of sizing error, as will be discussed below. (2) Aerosol is assumed to be composed of a single component with properties representative of ambient organic aerosol.
Organic compounds are the only, besides NH4NO3, semivolatile species in ambient aerosol. Ambient organic aerosol is composed of thousands of compounds of widely varying volatility (Robinson et al. Citation2007; Cappa and Jimenez Citation2010). Modeling all possible volatility distributions of ambient organic aerosol is impractical; a single component approximation is, therefore, used to provide a generally valid estimate of sizing error when measuring organic aerosol. The error when measuring organic mixtures should be bound between that corresponding to the highest and the lowest volatilities in a given mixture. The exact error for any given mixture composition can be calculated using Equation (Equation10[10] ).
The molecular weight of a surrogate organic compound was taken to be Mw = 200 g/mol (Kiss et al. Citation2003; Saleh and Khlystov Citation2009), the density ρ = 1.3 g/cm3, the gas phase diffusion coefficient D = 0.05 cm2/s, surface tension σ = 0.2 ergs/cm2 (Tao and McMurry Citation1989; Saleh et al. Citation2009), and the evaporation coefficient α = 0.1 (Saleh et al. Citation2009, Citation2012; Cappa and Jimenez Citation2010). If one assumes α = 1, the error will be approximately equal to that calculated for compounds with ten-fold higher volatility. For example, at α = 1 error for Cs = 1 μg/m3 will be similar in magnitude to those for Cs = 10 μg/m3 and α = 0.1.
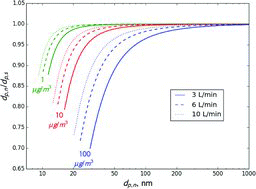
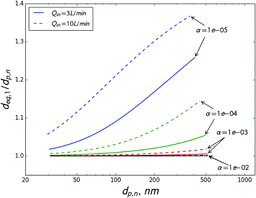
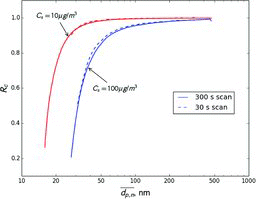
shows the ratios of nominal diameter (dp, n) to sampled diameter (dp, s) as a function of dp, n when measuring volatile aerosols with a single TSI long DMA at 3 L/min sheath flow. Results for compounds with volatilities (Cs) ranging from 0.1 to 100 μg/m3 are shown. This ratio is a measure of sizing error. Since volatile aerosols are consistently undersized, the nominal size is always smaller than the actual size of sampled particles. However, for compounds with Cs ⩽ 10 μg/m3, the error is less than 5% for particles larger than 30 nm. For compounds with Cs = 100 μg/m3, the error is larger, but is within 5% for particles larger than 85 nm. The figure also shows dp, n/dp, s as calculated for the scanning mode of operation. In general, the error for 300 s scans is very close to that in the constant voltage mode. A faster, 30 s scan produces larger error, especially for higher volatility compounds and smaller particle sizes.
According to gas/particle partitioning theory, ambient semivolatile organic aerosol is mostly composed of compounds with Cs that is less than or equal to the total organic aerosol concentration (Donahue et al. Citation2006). Since ambient organic aerosol concentrations are on the order of 1 to 10 μg/m3, the Cs of most compounds in ambient particles is lower than these values. This means that when measuring ambient aerosol error is expected to be fairly small. Since ambient aerosol is composed of a mixture of compounds of different volatilities, error would probably be even smaller, since effective Cs decreases during evaporation due to loss of higher volatility compounds.
At higher flow rates, errors are even smaller. shows the dp, n/dp, s ratio modeled for different sheath flow rates and the constant voltage mode. For example, at a sheath flow of 10 L/min and Cs ⩽ 10 μg/m3, the predicted error in the sampled size is less than 5% for particles larger than 20 nm. For Cs = 100 μg/m3 error is smaller than 5% for particles larger than 45 nm.
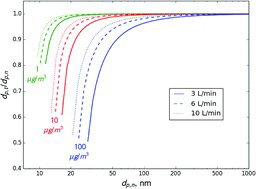
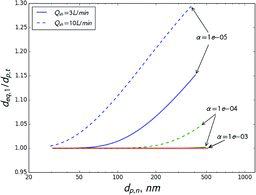
Sizing error for transferred particles is somewhat larger than that for sampled particles (). In other words, the size of transferred particles is overestimated to a larger extent than the sampled size is underestimated (dp, t/dp, n < dp, n/dp, s). However, the difference in error becomes significant only at ratios below 0.95; otherwise errors in sampled and transferred sizes are comparable in magnitude.
It should be stressed that the calculations presented in Figures – are an upper estimate of error when measuring organic semivolatile aerosols. The calculation assumes that sheath air is vapor-free, which may not be the case if sheath air is sampled from the same source as the particles. The assumed conditions could occur if: (1) sheath air is not recirculated and is taken from a different source than the sample air, (2) there is a significant temperature difference between the DMA and sample air, or (3) sampled aerosol is reactive with materials used in the DMA (stainless steel, plastic in the tubing, filter material, etc.). If care is taken to avoid such conditions, errors will be smaller.
For example, if sheath air is recirculated and the compound of interest is inert, the vapor concentration in sheath air will be very close to that in sample air, suppressing evaporation. This is because the vapor concentration in sheath air will gradually build up due to mixing in of the sample air. All of the sample air is removed from the DMA with the “excess” air, which is then filtered and recirculated to be supplied to the DMA as the sheath air. Sheath air should be equilibrated with the sample air within about 30 min. Since most organic compounds probably will not react with the DMA or the tubing material in the sheath line, the vapor concentration will quickly reach that of the sample air. Therefore, provides worst case scenario for ambient aerosol measurements. However, sheath air should be either recirculated or taken from the same source as sampled aerosol. Affinity of the compound(s) in question to the materials used in the DMA should be investigated to avoid significant errors. Care should also be taken when measuring very volatile aerosols (Cs > 10 μg/m3), especially smaller than 100 nm at low sheath flow rates. To avoid or minimize errors when measuring particles smaller than 100 nm, it is preferable to use higher sheath flow rates.
5.4 Using DMA-Selected Ammonium Nitrate Aerosol for Instrument Calibration
The above arguments for overestimation of errors do not apply to measurements of NH4NO3 aerosol, as is apparent from the experimental data discussed previously. Clearly, due to the high affinity of nitric acid to metal and other surfaces, it is efficiently removed in the DMA. This is supported by the very small effect that gaseous ammonia concentration has on observed sizing deviations as was discussed in Section 5.2 ().
Since NH4NO3 aerosol is used to calibrate ionization efficiency of Aerodyne aerosol mass spectrometers, calculations were made to estimate errors in the mass of the DMA-selected aerosol. shows the predicted ratio of transferred ammonium nitrate volume (and thus mass) to that expected based on the settings of the DMA at 3 L/min sheath flow at different temperatures. The estimated ratios when selecting dp, n = 300 nm NH4NO3 aerosol for different temperatures and sheath flows are summarized in . 300 nm aerosol is recommended for calibrating ACSM and AMS instruments (Allan et al. Citation2003). Error at this size is generally very small, the largest being about 5% at 25°C and 3 L/min sheath flow. This suggests that higher sheath flow rates and lower temperatures result in smaller errors. Thus, it is recommended that calibration is performed using higher sheath flow rates and, if possible, in cooler conditions.
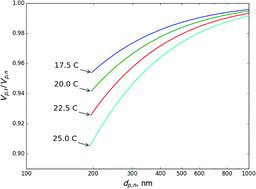
Table 2 Estimated ratio of the transmitted to set mass when selecting 300 nm ammonium nitrate aerosol using a single DMA
5.5 Errors when Measuring Hygroscopic Aerosols
It has been recognized for some time that hygroscopic particles can grow within DMAs. For example, when particle growth is slow due to constraints on water condensation, particles can continue growing in the second DMA of a TDMA system (Chuang Citation2003). Particle deliquescence and growth in the second DMA of TDMA systems was observed when the operating RH of the system is close to the deliquescence or the efflorescence RH of the sampled aerosol (Mikhailov et al. Citation2004; Biskos et al. Citation2006).
Measurements of hygroscopic aerosols when there are differences in RH between sample and sheath air represent a special case. Water is a very volatile compound (Cs = 1.7107 μg/m3 at 20°C). Therefore, its evaporation or condensation is very fast. Time needed to achieve equilibrium at a new RH can be as short as a few milliseconds and is typically less than 0.1 s (Kerminen Citation1997) unless there are significant kinetic constraints due to surface coatings on the droplet surface, in which case tens of seconds could be required to achieve equilibrium (Chuang Citation2003; Chan and Chan Citation2005; Sjogren et al. Citation2007).
In Section 2 it was shown that the average electrical mobility of a particle during its passage through the DMA classification region determines whether or not the particle is selected by the DMA (Equation (Equation10[10] )). Assuming that there are no significant evaporation or condensation constraints, the evaporation or growth time (less than 0.1 s) is significantly shorter than the residence time in the DMA (3 s or more). This implies that the average mobility during the time a particle spends in the DMA is very close to the particle mobility at equilibrium with sheath air. Thus, to a first approximation, the DMA will report a size corresponding to the equilibrium size at the sheath RH.
The range of applicability of this approximation was investigated by using (NH4)2SO4 aerosol as an example. The aerosol was assumed to be initially in a wet metastable state in equilibrium with air at 40% RH. Sheath air RH was set to 90%. Particle growth was modeled using Equation (Equation8[8] ) to obtain the particle size as a function of time. The expressions for water vapor activity, which is needed for calculation of water vapor concentration at the particle surface, and the density of (NH4)2SO4 solutions as a function of molality of (NH4)2SO4 were taken from Tang and Munkelwitz (Citation1994). If aerosol is assumed to be initially dry instead of being in a metastable state, its growth is somewhat faster. This is because the initial driving force for water condensation is stronger due to a larger initial difference in water vapor concentration between the particle surface, which in this case is zero initially, and the surrounding air. The calculations were repeated for several initial sizes and the calculated dp(t) was used to determine the nominal size for each initial particle size using Equation (Equation10
[10] ).
shows the ratio of the equilibrium size (deq, 1) at the sheath RH to the nominal size (dp, n) for different values of accommodation coefficient (α). Deviation of deq, 1 from dp, n was found to be negligible for α > 0.01. The size of transferred particles (dp, t) is virtually identical to deq, 1 for α > 10− 3 (). The deviations of dp, n and dp, t from deq, 1 increase at larger dp, n. This is because larger particles equilibrate slower than smaller particles. The deviations are larger for lower values of α for the same reason—the equilibration is slower at lower α values. It is also interesting to note that deviations are larger for higher sheath flow rates. When equilibration time is much shorter than tf, the integral in Equation (Equation10[10] ) is approximately equal to tfZ(deq, 1). As equilibration time becomes comparable to tf, deviation of the integral from this value increases. Therefore, these deviations are larger at high sheath flow rates than at low flow rates.
To summarize, the equilibrium size at the RH of sheath flow is a good approximation for dp, n and dp, t when α > 0.01, especially at low sheath flow rates.
5.6 Transfer and Selection Functions
As was discussed in Section 2, the size of volatile particles sampled by a DMA is different from their size at the exit of the classifier. Therefore, an additional function needs to be defined for sampled particles, Ωs(dp). This function defines the probability that a particle with size at the entrance of the DMA of dp will be sampled and transferred through the DMA at the given conditions. Likewise, the transfer function, Ωt(dp), is defined as the probability of a particle with size at the exit of the DMA of dp to be transferred through the DMA at the given conditions. For nonvolatile particles, Ωs is identical to Ωt because stable particles do not change size in the DMA.
The effect of particle evaporation on the DMA transfer and selection functions in constant-voltage mode of operation is shown in . The figure shows Ωs and Ωt modeled for NH4NO3 aerosol for a sheath flow rate of 3 L/min and 25°C. Sample and monodisperse flow rates were taken to be 0.3 L/min.
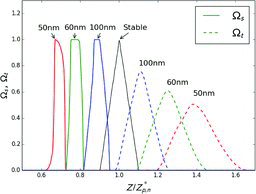
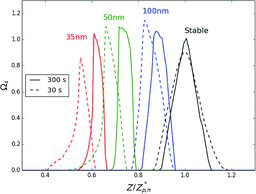
The selection function becomes progressively narrower as the centroid (mean) nominal size decreases. Ωs attains a trapezoidal form with a maximum value of 1. However, below 60 nm, the form of Ωs becomes distorted, being concave on the low mobility side and convex on the high mobility side. Unlike Ωs, Ωt becomes progressively broader as nominal size decreases. At 50 nm nominal size it is approximately two times broader than the width of the transfer function of a stable aerosol. Despite getting broader, the shape of Ωt remains fairly symmetrical. These observations follow the trend observed in NH4NO3 measurements ().
shows Ωs modeled for the scanning mode of operation. Because the scanning mode is used only to measure aerosol size distributions, Ωt is not considered here. NH4NO3 aerosol at 25°C was used as an example. The sheath flow rate was taken to be 3 L/min. Calculations were done for two scan durations, representative of the most commonly used fast and slow scan rates, 30 s and 300 s. DMA voltage was exponentially scanned from 10 V to 10,000 V. The figure also shows Ωs for a stable aerosol.
Similar to the constant voltage mode, Ωs in the scanning mode becomes progressively narrower and distorted as the centroid nominal size decreases. The shape, however, does not attain a trapezoidal form. An interesting result is that the maximum value of Ωs in the scanning mode often exceeds 1. This indicates that, due to particle evaporation, the flux of certain size particles through monodisperse flow could be higher than the flux of these particles through sample flow. Both the shape distortion and higher values of Ωs are more pronounced at the higher scan rate.
To explain these observations, it should be recalled that the scanning mode is an inherently time-based classification process. The time since the start of the scan determines the mean electrical field experienced by particles in the DMA and thus the centroid mobility of the selected particles. If the flow in the DMA were radially uniform, there would be a clear correspondence between the centroid mobility of selected particles and the time at which they exit the DMA (Wang and Flagan Citation1990). However, due to radial variation of gas velocity, particles entering the DMA at different times and different radial positions can experience different electrical field histories. This leads to distortion of the transfer function and a shift of the centroid mobility from the expected “ideal” value. These effects are especially pronounced at large (Collins et al. Citation2004). In fact, distortion of the transfer function of stable particles modeled in this study for a 30 s scan is very close to that reported by Collins et al. (Citation2004).
Particle evaporation exacerbates this nonideal behavior. Due to differences in particle trajectories, at any given time the DMA could select particles that have the same initial size, but that have spent different times in the DMA. In other words, particles that have the same initial size but that have entered the DMA at different times could exit the DMA simultaneously. As volatile particles change their size in the DMA, their trajectories under a varying electrical field become even more dependent on the radial position and time at which they enter the DMA. Because of this, particles within an even wider range of entrance times could be selected by (i.e., exit) the DMA at any given moment. This could lead to a flux of a given initial size particles through the monodisperse flow to be greater than their flux into the DMA. This leads to a Ωs larger than 1 for these particles.
Changes in Ωs due to particle evaporation or growth could lead to significant errors in size distribution measurements. Aerosol size distributions are measured by changing the DMA voltage, either in discrete steps or by exponentially ramping it, and counting particles selected by the DMA at each nominal size. As was discussed in the previous sections, dp, s ≠ dn, p for volatile particles. Therefore, volatile particles are either undersized if they evaporate, or oversized if they grow. The sizing error can be significant, especially for high volatility compounds and at small particle sizes. Another kind of error arises due to improper charge correction. Since DMAs select only charged particles, measured particle counts need to be corrected for size-dependent charging efficiency to obtain total particle concentration at each nominal size. Charge correction depends strongly on particle size (Wiedensohler Citation1988). Therefore, sizing errors cause errors in measured aerosol concentrations. Deviations of the shape of Ωs from its conventional shape can also lead to errors during size distribution inversion.
To estimate the relative importance of these two effects (counting correction and the shape of Ωs), the following ratio was calculated:
[22] in which f is the size-dependent fraction of singly charged particles, Ωs, n is Ωs for stable particles,
is the centroid nominal size and
is the mean initial size of volatile particles that would be selected at each given moment during a scan. Doubly charged particles are ignored in this analysis for simplicity. Rc approximates the net result of the two effects on the derived particle concentration at each
by assuming that the aerosol size distribution does not vary with size. While such distributions are never encountered in practice, the error for each given distribution can be approximated by multiplying Rc by the ratio of the value of the size distribution at
to its value at
. The exact error can be obtained only by inverting the measured particle count data using Ωs. It is not feasible to do so for every possible aerosol size distribution; therefore, only Rc is considered here to provide an estimate of the counting error.
shows Rc as a function of for evaporating particles. The error due to charging correction should lead to an overestimation of particle counts for particle diameters smaller than 125 nm, because
and the fraction of singly charged particles increases up to this size (Wiedensohler Citation1988). However, the calculated Rc is always smaller than 1. This is because the fraction of selected volatile particles, which is determined by Ωs, is smaller than that of stable particles. While the maximum value of Ωs could be often larger than 1, its shape is narrower than that of Ωs, n resulting in a smaller selected fraction for volatile particles. This reduction in selection efficiency is larger than the overcounting due to improper charging correction, resulting in Rc < 1.
6 CONCLUSIONS
A theoretical analysis of the effect of particle size changes during the classification process on the DMA sizing accuracy has been presented. It is shown that for an evaporating or growing particle to be classified by a DMA at given operating conditions (sheath flow rate and DMA voltage) its average mobility during classification should be equal to that of a stable particle that would be selected under the same operating conditions. This implies that evaporating particles will be always undersized by DMAs and that the size of transmitted particles will be smaller than the size set by the DMA. Likewise, growing particles will be oversized and transmitted particles will be larger than the set size.
This theory agrees well with measurements of NH4NO3 aerosol and results of numerical modeling of particle growth or evaporation in DMAs. The model was used to predict sizing error when measuring semivolatile aerosols. The predicted error increases with compound volatility and is especially pronounced for particles smaller than 100 nm. The upper estimates of sizing errors were predicted to be fairly small for compounds with saturation vapor concentration below 10 μg/m3, assuming an accommodation/evaporation coefficient is 0.1. However, care should be taken when measuring higher volatility aerosols, especially for particles smaller than 100 nm at low sheath flow rates.
To minimize error, higher sheath flow rates should be used and sheath air should be either recirculated or taken from the same source as sampled aerosol. Sizing errors cannot be avoided when measuring semivolatile components, such as NH4NO3, that can react with or deposit to surfaces in the DMA. To minimize sizing error when working with such compounds, higher sheath flow rates should be used and, if possible, measurements should be done at lower temperatures to suppress compound volatility.
When measuring hygroscopic aerosol, equilibrium particle size at the RH of sheath flow is a good approximation for the size that will be reported and transmitted by the DMA, if there are no kinetic constraints for hygroscopic growth (accommodation coefficient of water larger than 0.01).
Numerical modeling shows that sizing error is generally larger for the scanning mode than in the constant-voltage mode. Error is larger when faster scans are used. In constant-voltage mode, the selection function becomes progressively narrower as the centroid nominal size decreases. Below 60 nm nominal size, the selection function becomes distorted, being concave at low mobility and convex at high mobility. On the other hand, the transfer function becomes progressively broader as the nominal size decreases, but remains fairly symmetrical. Similar to the constant-voltage mode, the selection function in the scanning mode becomes progressively narrower and distorted as the centroid nominal size decreases. The shape, however, does not attain a trapezoidal form. Due to distortion of the selection function, the concentration of evaporating particles is predicted to be underestimated, especially at small sizes and high aerosol volatility.
ACKNOWLEDGMENTS
The author is most grateful to Dr. Arja Even and Anne Toivonen for performing most of the measurements reported here and Dr. Harry ten Brink for fruitful discussions during the experimental part of the study. The help of Rene Otjes with ammonia measurements is also greatly appreciated.
FUNDING
A part of the material is based upon work supported by the National Science Foundation under grant no. AGS-09-55845. The author is also grateful to the Department of Economic Affairs of the Netherlands for their support during the experimental part of this study under contract no. 53478).
NOMENCLATURE
| Cc | = | Cunningham slip correction factor |
| Cs | = | mole-fraction-weighed saturation vapor concentration of a mixture; for a single component aerosol it is equal to its saturation vapor concentration |
| Csh | = | total concentration of vapors in the sheath air |
| D | = | diffusion coefficient of vapor molecules |
| dp, s | = | particle diameter at the entrance of the DMA classification region (sampled diameter) |
| = | mean particle diameter at the entrance of the DMA classification region | |
| dp, t | = | particle diameter at the exit of the the DMA classification region (transferred diameter) |
| deq, 1 | = | particle diameter at equilibrium at the relative humidity of the sheath air |
| dp | = | particle diameter |
| dp, n | = | nominal diameter of a particle; it is equal to the diameter of a stable singly charged particle that is selected by the DMA at the given voltage (or time from the beginning of the scan) and sheath flow rate |
| = | mean (centroid) nominal diameter | |
| F | = | Fuchs–Sutugin correction factor |
| e | = | elementary charge |
| E1 | = | electrical field at the surface of the inner cylinder of the DMA |
| E1, 0 | = | electrical field at the surface of the inner cylinder of the DMA at the start of a scan |
| Er | = | radial electrical field in the DMA |
| i | = | number of elementary charges on a particle |
| k | = | = r1/r2 |
| Kn | = | Knudsen number |
| L | = | length of the DMA classification region |
| Qa | = | sample air flow rate |
| Qsh | = | sheath flow rate |
| r | = | radial coordinate |
| r1 | = | radius of the inner cylinder of the DMA |
| r2 | = | radius of the outer cylinder of the DMA |
| rin | = | radial position at which a particle enters the DMA |
| t | = | time |
| = | = ts/τ | |
| tf | = | mean gas residence time in the classification region of the DMA |
| ts | = | scan time |
| u | = | gas radial velocity |
| uav | = | average gas velocity in the DMA |
| V | = | voltage applied to the DMA |
| x | = | axial coordinate |
| y | = | = r/r2 |
| Vp, n | = | nominal aerosol volume |
| Vp, t | = | transferred aerosol volume |
| Zp | = | particle electrical mobility |
| = | mean particle mobility in the classification region of the DMA | |
| Zp, n | = | nominal particle electrical mobility. It is equal to the mobility of a stable particle that is selected by the DMA at the given voltage (or time from the beginning of the scan) and sheath flow rate |
| Z*p, n | = | centroid nominal particle electrical mobility |
| α | = | accommodation or evaporation coefficient |
| σ | = | surface tension |
| λa | = | mean free path of air molecules |
| μ | = | gas viscosity |
| Ωs | = | sampling function |
| Ωt | = | transfer function |
| ρ | = | particle density |
| τ | = | scan time constant |
SUPPLEMENTAL MATERIAL
Supplemental data for this article can be accessed on the publisher's website.
am_Supplement_print.zip
Download Zip (124.9 KB)REFERENCES
- Allan, J., Jimenez, J., Williams, P., Alfarra, M., Bower, K., Jayne, J., et al. (2003). Quantitative Sampling Using an Aerodyne Aerosol Mass Spectrometer - 1. Techniques of Data Interpretation and Error Analysis. J. Geophys. Res. - Atmos., 108:4090, doi:10.1029/2002JD002358.
- Allen, M., and Raabe, O. (1985). Slip Correction Measurements of Spherical Solid Aerosol-Particles in an Improved Millikan Apparatus. Aerosol Sci. Technol., 4:269–286.
- Bird, R., Stewart, W., and Lightfood, E. (1960). Transport Phenomena. Wiley, New York.
- Biskos, G., Paulsen, D., Russell, L.M., Buseck, P.R., and Martin, S.T. (2006). Prompt Deliquescence and Efflorescence of Aerosol Nanoparticles. Atmos. Chem. Phys., 6:4633–4642.
- Cappa, C.D., and Jimenez, J.L. (2010). Quantitative Estimates of the Volatility of Ambient Organic Aerosol. Atmos. Chem. Phys., 10:5409–5424.
- Chan, M., and Chan, C. (2005). Mass Transfer Effects in Hygroscopic Measurements of Aerosol Particles. Atmos. Chem. Phys., 5:2703–2712.
- Chuang, P. (2003). Measurement of the Timescale of Hygroscopic Growth for Atmospheric Aerosols. J. Geophys. Res. - Atmos., 108: doi:10.1029/2002JD002757.
- Collins, D., Cocker, D., Flagan, R., and Seinfeld, J. (2004). The Scanning DMA Transfer Function. Aerosol Sci. Technol., 38:833–850.
- Dassios, K., and Pandis, S. (1999). The Mass Accommodation Coefficient of Ammonium Nitrate Aerosol. Atmos. Environ., 33:2993–3003.
- Donahue, N., Robinson, A., Stanier, C., and Pandis, S. (2006). Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics. Environmen. Sci. Technol., 40:2635–2643.
- Fissan, H., Helsper, C., and Thielen, H. (1983). Determination of Particle Size Distribution by Means of an Electrostatic Classifier. J. Aerosol Sci., 14:354–357.
- Hagwood, C., Sivathanu, Y., and Mulholland, G. (1999). The DMA Transfer Function with Brownian Motion a Trajectory/Monte-Carlo Approach. Aerosol Sci. Technol., 30:40–61, doi:10.1080/027868299304877.
- Kerminen, V.M. (1997). The Effects of Particle Chemical Character and Atmospheric Processes on Particle Hygroscopic Properties. J. Aerosol Sci., 28:121–132.
- Kiss, G., Tombacz, E., Varga, B., Alsberg, T., and Persson, L. (2003). Estimation of the Average Molecular Weight of Humic-Like Substances Isolated from Fine Atmospheric Aerosol. Atmos. Environ., 37:3783–3794.
- Knutson, E., and Whitby, K. (1975). Aerosol Classification by Electric Mobility: Apparatus, Theory and Applications. J. Aerosol Sci., 6:443–451.
- Knutson, E.O. (1976). Extended Electric Mobility Method for Measuring Aerosol Particle Size and Concentration, in Fine Particles: Aerosol Generation, Measurement, Sampling, and Analysis, B. Liu, ed., Academic Press, New York, pp. 739–762.
- Kousaka, Y., Okuyama, K., Adachi, M., and Mimura, T. (1986). Effect of Brownian Diffusion on Electrical Classification of Ultrafine Aerosol Particles in Differential Mobility Analyzer. J. Chem. Eng. Japan, 19:401–407, doi:10.1252/jcej.19.401.
- Mikhailov, E., Vlasenko, S., Niessner, R., and Poschl, U. (2004). Interaction of Aerosol Particles Composed of Protein and Salts with Water Vapor: Hygroscopic Growth and Microstructural Rearrangement. Atmos. Chem. Phys., 4:323–350.
- Mozurkewich, M. (1993). The Dissociation Constant of Ammonium Nitrate and its Dependence on Temperature, Relative Humidity and Particle Size. Atmos. Environ., 27A:261–270.
- Neuman, J., Huey, L., Ryerson, T., and Fahey, D. (1999). Study of Inlet Materials for Sampling Atmospheric Nitric Acid. Environ. Sci. Technol., 33:1133–1136.
- Rader, D., and McMurry, P. (1986). Application of the Tandem Differential Mobility Analyzer to Studies of Droplet Growth or Evaporation. J. Aerosol Sci., 17:771–787.
- Robinson, A., Donahue, N.M., Shrivastava, M., Weitkamp, E., Sage, A., Grieshop, A., et al. (2007). Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging. Science, 315:1259–1262.
- Saleh, R., and Khlystov, A. (2009). Determination of Activity Coefficients of Semi-Volatile Organic Aerosols Using the Integrated Volume Method. Aerosol Sci. Technol., 43:838–846.
- Saleh, R., Shihadeh, A., and Khlystov, A. (2009). Determination of Evaporation Coefficients of Semi-Volatile Organic Aerosols Using an Integrated Volume-Tandem Differential Mobility Analysis (IV-TDMA) Method. J. Aerosol Sci., 40:1019–1029.
- Saleh, R., Khlystov, A., and Shihadeh, A. (2012). Determination of Evaporation Coefficients of Ambient and Laboratory-Generated Semivolatile Organic Aerosols from Phase Equilibration Kinetics in a Thermodenuder. Aerosol Sci. Technol., 46:22–30.
- Seinfeld, J., and Pandis, S.N. (2006). Atmospheric Chemistry and Physics: From Air Pollution to Climate Change(2nd ed.). Wiley & Sons, Inc., Hoboken, New Jersey.
- Sjogren, S., Gysel, M., Weingartner, E., Baltensperger, U., Cubison, M.J., Coe, H., et al. (2007). Hygroscopic Growth and Water Uptake Kinetics of Two-Phase Aerosol Particles Consisting of Ammonium Sulfate, Adipic and Humic Acid Mixtures. J. Aerosol Sci., 38:157–171, doi:10.1016/j.jaerosci.2006.11.005.
- Stolzenburg, M.R. (1988). An Ultrafine Aerosol Size Distribution Measuring System. Ph.D. thesis, Mechanical Engineering Department, University of Minnesota, Minneapolis.
- Swietlicki, E., Hansson, H.C., Hameri, K., Svenningsson, B., Massling, A., McFiggans, G., et al. (2008). Hygroscopic Properties of Submicrometer Atmospheric Aerosol Particles Measured with H-TDMA Instruments in Various Environments - A Review. Tellus Series B - Chem. Phys. Meteorol., 60:432–469, doi:10.1111/j.1600-0889.2008.00350.x.
- Tang, I., and Munkelwitz, H. (1994). Water Activities, Densities, and Refractive Indexes of Aqueous Sulfates and Sodium Nitrate Droplets of Atmospheric Importance. J. Geophys. Res. - Atmos., 99:18801–18808.
- Tao, Y., and McMurry, P. (1989). Vapor Pressure and Surface Free Energies of C14 C18 Monocarboxylic Acids and C5 and C6 Dicarboxylic Acids. Environ. Sci. Technol., 23:1519–1523.
- ten Brink, H., Plomp, A., Spoelstra, H., and van de Vate, J. (1983). A High Resolution Electrical Mobility Aerosol Spectrometer (MAS). J. Aerosol Sci., 14:589.
- ten Brink, H., Khlystov, A., Kos, G., Tuch, T., Roth, C., and Kreyling, W. (2000). A High-Flow Humidograph for Testing the Water Uptake by Ambient Aerosol. Atmos. Environ., 34:4291–4300.
- Twomey, S. (1977). Introduction to the Mathematics of Inversion in Remote Sensing and Indirect Measurements. Dover Publications, Inc., Mineola, NY.
- Wang, S., and Flagan, R. (1990). Scanning Electrical Mobility Spectrometer. Aerosol Sci. Technol., 13:230–240.
- Wiedensohler, A. (1988). An Approximation of the Bipolar Charge-Distribution for Particles in the Sub-Micron Size Range. J. Aerosol Sci., 19:387–389, doi:10.1016/0021-8502(88)90278-9.
- Winkelmayr, W., Reischl, G., Linde, A., and Berner, A. (1991). A New Electromobility Spectrometer for the Measurement of Aerosol Size Distributions in the Size Range from 1 to 1000 nm. J. Aerosol Sci., 22:289–296.
- Wyers, G., Otjes, R., and Slanina, J. (1993). A Continuous-Flow Denuder for the Measurement of Ambient Concentrations and Surface-Exchange Fluxes of Ammonia. Atmos. Environ., 27A:2085–2090.