
ABSTRACT
We have investigated the vapor wall loss of semi-volatile organic compounds (SVOCs) in a Teflon smog chamber. We studied the vapor wall loss of seven SVOCs with known saturation concentrations, including alkanes (hexacosane, pentacosane, docosane, eicosane, and d62-squalane), an organic acid (oleic acid), and a polyol (levoglucosan) in single-component and binary-component (organic) systems, using ammonium sulfate (AS) seeds to constrain the particle wall loss. We coated inorganic particles with SVOCs and measured the loss of organics from those particles to constrain the wall losses, observing loss rates proportional to the saturation concentrations of the SVOCs. The loss rate of oleic acid mixed with d62-squalane was proportional to its mole fraction in the mixture. Our results show that the vapor wall-loss rates of SVOCs are significant, quasi-irreversible, and proportional to the SVOC vapor concentrations. The vapor wall-loss rate constant of the SVOCs that we studied in the CMU chamber is 3.8 ± 0.3 h−1; this is comparable to values in other chambers with similar surface area to volume ratios. Our results are also consistent with a relatively high mass accommodation coefficient for SVOCs, αorg > 0.1.
© 2016 American Association for Aerosol Research
EDITOR:
Introduction
Teflon smog chambers have been extensively used to study the formation, physical properties, and ongoing chemical reactions of secondary organic aerosol (SOA; Stern et al. Citation1987; Pandis et al. Citation1991; Zhang et al. Citation1992; Hoffmann et al. Citation1997; Odum et al. Citation1997; Presto et al. Citation2005ab). More than two decades of research supports an understanding that SOA contains a high fraction of semi-volatile organic compounds (SVOCs; Pankow Citation1994; Odum et al. Citation1996; Kamens et al. Citation1999; Donahue et al. Citation2006) though discovery of oligomers in SOA (Kalberer et al. Citation2004) and more recently extremely low volatility organic compounds (ELVOCs) formed in the gas phase (Ehn et al. Citation2014; Kokkola et al. Citation2014) have called into question what fraction of SOA formation involves SVOCs. Conversely, POA was long treated as being non-volatile, but extensive evidence has shown that a large fraction of POA consists of SVOCs (Robinson et al. Citation2007; Grieshop et al. Citation2009). Once again, Teflon chambers have been an important tool used to probe POA volatility and the evolution of POA due to photochemical aging (Weitkamp et al. Citation2007; Sage et al. Citation2008; Chirico et al. Citation2010).
The interaction between SVOC vapors and Teflon chamber walls is a key confounding factor influencing organic-aerosol behavior in chamber experiments. The loss of organic vapors to the Teflon chamber walls reduces the condensation of vapors to suspended particles and thus reduces the apparent SOA mass yield. Furthermore, if vapor wall losses are selective, this can bias the chemical composition and properties of the formed SOA. Direct treatment of particle wall loss is straightforward (Stanier et al. Citation2008). Recently, more attention has been given to the vapor wall loss due to the condensation of organic vapors onto or into Teflon chamber walls (McMurry and Grosjean Citation1985; Loza et al. Citation2010; Matsunaga and Ziemann Citation2010; McVay et al. Citation2014; Yeh and Ziemann Citation2014; Zhang et al. Citation2014, Citation2015). Loss measurements have been reported for VOC vapors, including n-alkanes, 1-alkenes, 2-alcohols, and 2-ketones, and the results have been interpreted in terms of reversible sorption into Teflon chamber walls (Matsunaga and Ziemann Citation2010). Other studies have shown that 2,3-epoxy-1,4-butanediol, and glyoxal have similar wall-loss behavior (Loza et al. Citation2010), and that organonitates show significant vapor wall loss again consistent with reversible partitioning (Yeh and Ziemann Citation2014). It may be that apparent SOA yields can be biased low by a factor of four due to vapor wall loss (Zhang et al. Citation2014).
Our goal is to quantify the loss of SVOC vapors to the Teflon chamber walls. Matsunaga and Ziemann observed VOCs present in the gas phase at equilibrium (Matsunaga and Ziemann Citation2010). By design, they studied losses for relatively volatile compounds well below saturation. This was to avoid both suspended particle formation and condensation to the walls driven solely by nucleation of a condensed phase on the walls. Broadly, the compounds they studied can be classified as intermediate volatility organic compounds (IVOCs). They injected a carefully quantified aliquot of various IVOCs into an otherwise empty chamber, waited for the chamber to become well mixed, and then measured the gas-phase concentration. They observed a deficit in the gas phase inversely correlated with vapor pressure but also related to the IVOC molecular structure. Furthermore, they were able to recover a fraction of any missing materials by evacuating the chamber and re-filling it with clean air. They concluded that the results were consistent with organic vapors reaching an absorptive equilibrium within a thin layer of disordered polymer at the Teflon surface. Though the organics presumably have a very high activity coefficient for sorption within the Teflon matrix, the extremely large overall mass of this layer (compared with 10–100 µg m−3 levels of organics in SOA conditions) resulted in significant mass uptake for the IVOCs, consistent with a sorptive equilibrium given by[1] where Fwall is the fraction of organics lost to the chamber walls, C° is the saturation concentration of the IVOCs, and Cwall is an effective wall concentration for partitioning. Broadly, they found 2 mg m−3 < Cwall < 24 mg m−3 in a 6 m3 chamber, depending on structure.
The Matsunaga and Ziemann findings can be extrapolated to SVOCs. The implication is that the large majority of SVOCs will reside in the wall at equilibrium. For example, when considering Cwall = 10 mg m−3, an SVOC with C* ∼ 10 µg m−3 would have Fwall ≈ 0.999. Thus, 99.9% of the SVOC vapor could be lost to the Teflon walls, and the vapor loss of SVOCs to the chamber walls may be quasi-irreversible.
There have been relatively few observations to directly constrain the behavior of SVOCs in chambers. Zhang et al. used chemical ionization mass spectrometry (CIMS) to track the concentration of 25 oxidized organic compounds generated from the photooxidation of SOA precursors (Zhang et al. Citation2015), but reported data after an hour or so of SOA formation. Yeh and Ziemann monitored the vapor loss of a suite of synthesized alkyl nitrates to constrain the loss of alkyl nitrate products from C8−C14 n-alkanes reacted with OH radicals in the presence of NOx (Yeh and Ziemann Citation2014).
The objective of this study is to map the vapor wall-loss behavior of SVOCs with well-known model compounds. We probe SVOC vapor wall loss through the measurement of suspended inorganic particles coated with SVOCs. Our objective is to establish a gas-particle equilibrium at saturation conditions where the wall is a minor perturbation and then to follow the mass loss from the particles. The well-established measurement methods for particles allow us to measure the loss of SVOC vapors for a range of molecules. Here, we present results for seven SVOCs with known saturation concentrations, including alkanes (hexacosane, pentacosane, docosane, eicosane, and d62-squalane), a carboxylic acid (oleic acid), and a polyalcohol (levoglucosan) in single-component and binary-component (organic) systems.
Methods and materials
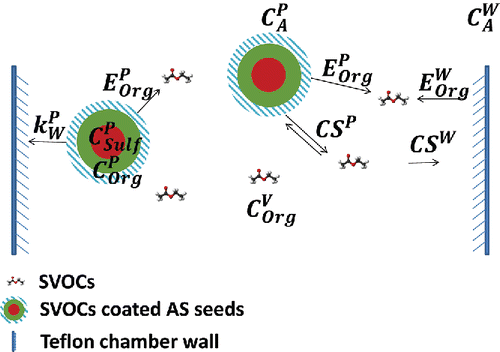
Our experimental design is conceptually straightforward, as shown in . We measure the mass loss from suspended particles to infer the vapor wall loss. We coat ammonium sulfate seeds with an SVOC in a Teflon chamber. The SVOCs establish an equilibrium between the gas phase and the condensed phase (on the ammonium sulfate particles) on a timescale equal to the suspended-particle condensation sink (roughly 1 min for these experiments; Saleh et al. Citation2013). At the same time, SVOC vapors are continuously lost to the chamber walls on a timescale equal to the wall condensation sink (roughly 10 min for the CMU chamber). SVOC vapor wall loss perturbs the system by reducing the vapor concentration and thus the SVOC activity in the gas phase (the saturation ratio); the suspended particles (with a pure condensed-phase activity of 1.0) then respond by evaporating SVOCs to re-establish the equilibrium. The mass loss of SVOCs from the suspended particles thus balances the SVOC vapor wall loss. Strictly speaking, the activity of the SVOC vapors remains in a steady state at 1 − ϵ so that there is a small thermodynamic driving force for evaporation from the suspended particles to counterbalance the driving force for vapor sorption into the Teflon walls. For our typical conditions, ϵ ≃ 0.1, and so we should measure a loss flux within 10% of the maximal value.
Scheme 1. The partitioning of SVOCs between the gas phase, particle phase, and Teflon chamber walls. Ammonium sulfate particles coated with organics sustain the SVOC vapors near their saturation concentration. The vapor wall loss of the SVOCs is thus balanced by the mass loss of organics from the suspended particles.
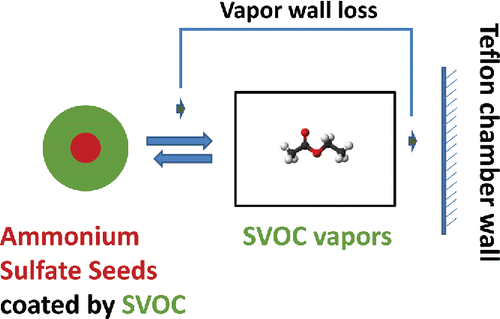
We can calculate the vapor wall loss from the mass decrease of SVOCs on the suspended particles; however, we need to know the absolute mass flux from the suspended particles (not the flux per particle) and so must consider particle wall losses. To first order, particle wall losses do not influence the suspension as they do not influence the activity (saturation ratio) of the SVOC in either phase but simply reduce the total amount of the condensed phase. Particle wall losses (typically with a timescale of hours) do lower the condensation sink and thus lengthen the equilibrium timescale, which becomes important once the condensation sink becomes similar to any vapor wall loss timescale as the steady-state vapor concentration will fall below the equilibrium vapor pressure over the suspended particles.
We conducted experiments with eight organics shown in . We performed the experiments in the Carnegie Mellon University (CMU) smog chamber, a 10 m3 Teflon bag suspended in a temperature-controlled room. Many details of the experimental procedures have been described previously (Hildebrandt et al. Citation2009), and so here we provide only a brief synopsis. Prior to each experiment, we cleaned the bag by flushing with clean, dry air and heating it to 35°C under UV irradiation. After cleaning, we usually maintained the chamber a constant temperature (with some exceptions described below). We used ammonium sulfate seed particles ((NH4)2SO4, 99.99%, Sigma Aldrich, St. Louis, MO, USA), which we formed by atomizing a 1 g L−1 (NH4)2SO4 solution in ultrapure deionized water to produce droplets that passed through a diffusion dryer and a neutralizer before they entered the chamber. In some cases, we mixed glucose in the solution with the ammonium sulfate. The seed concentrations were 2–3 × 104 particle cm−3, with a median diameter of ∼100 nm. We injected SVOCs into the chamber by gentle vaporization, after which they condensed onto the ammonium sulfate seeds with minimal nucleation. We used a small, resistive metal heater enclosed in a stainless-steel sheath to evaporate the organics inside the chamber, placing the organics in an indentation on the stainless-steel surface before inserting the heater into the chamber on the end of a long tube. With a flow of clean, dry dispersion air for mixing, we power-cycled the heater until the organics completely evaporated. The SVOC injection (18–50 µg m−3) lasted for 10–25 min. Particle number increases during the injection due to SVOC self-nucleation were in the range of 3–10%, causing a negligible change in the suspended-particle surface area (condensation sink) and thus minimally influencing the dynamics of the coated particles.
Table 1. Molecular formulas and thermodynamic properties of glucose, oleic acid, levoglucosan, hexacosane, pentacosane, docosane, eicosane, and squalane.
Our method is to measure any SVOC vapor wall loss by measuring the SVOC evaporation flux from the suspended particles. The loss of SVOCs from the particles causes them to shrink and most importantly decreases the ratio of SVOCs to the ammonium sulfate seeds. We measure the particle size distribution with a scanning mobility particle sizer (SMPS, TSI classifier model 3080, CPC model 3772 or 3010) and the mass concentrations and chemical composition of the particles with a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS, Aerodyne Research, Inc.; Drewnick et al. Citation2005). We thus constrain the total suspended particle volume, modal diameter, suspended mass, and the ratio of organics to sulfate (org:sulf) in the seeds as a function of aerodynamic diameter. The most precise measurement is org:sulf, but to constrain the overall evaporation flux we incorporate all of these measurements. We operate the HR-ToF-AMS according to the common protocol with the vaporizer temperature at 600°C. We collect mass spectra and particle time-of-flight (pToF) data in V-mode with a 1-min average.
We obtain single-particle mass spectra for particles with dva 180 nm using the light-scattering single-particle (LSSP) mode (Cross et al. Citation2009; Robinson et al. Citation2013), principally to measure the particle collection efficiency (CE). For all experiments, the HR-ToF-AMS alternates between V-mode and LSSP mode for 60 s each. The full design and operation of the LSSP module for the AMS have been described in full elsewhere (Cross et al. Citation2009). Briefly, LSSP allows the AMS to operate as a single-particle mass spectrometer. A continuous laser (405 nm, 50-mW; LC BCL-050-405; Crystal Laser) perpendicular to the particle beam in the time-of-flight chamber provides optical size and velocity for individual particles from scattered light signals for particles with dva
180 nm. Importantly, these light-scattering data are independent of the mass-spectrometer data. Each light-scattering event prompts selective acquisition of time-resolved mass spectra with a time resolution of 30 µs. Because acquisition is confined to chopper cycles where a particle is identified, the data load remains manageable. Thus, we obtain a measure for each particle of optical size and velocity (from the scattered light measurement) possibly a mass spectrum. However, not all particles generating a light-scattering signal also generate a mass spectrum, because CE is generally less than 1.0.
Particle bounce from the surface of the vaporizer is the dominant term causing CE < 1.0, provided that particles are spherical and within the transmission window of the aerodynamic lens (size range of 60–600 nm). Other loss terms can be important for non-spherical particles and those outside of the size range of the lens (Huffman et al. Citation2005), but do not apply to the particles measured in this study. We thus define CE as the fraction of particles identified by light scattering that generate a signal in the mass spectrometer of at least six ions. This is an operationally defined threshold for particles providing meaningful signal in the mass spectrometer (Liu et al. Citation2013). We include all particles with mass signals above the threshold, regardless of any delay in arrival time that may be attributable to particle bounce within the vaporization/ionization region.
In our experiments, the particles consist of an SVOC shell on a solid ammonium-sulfate core, and so CE evolves as the core-shell thickness changes due to evaporation (Robinson et al. Citation2015). We need to determine CE as a function of time to find the absolute evaporation rate from our AMS measurements, as described below. We have every reason to believe that CE evolves monotonically and slowly during our experiments, and so we use a linear fit of the raw CE vs. time data to smooth the observations.
Results and discussion
Observation of vapor wall loss of glucose and oleic acid
First, we shall consider one nearly non-volatile and one semi-volatile organic in detail: glucose and oleic acid. Glucose is an extremely low volatility organic compound (ELVOC) that barely exists in the gas phase. The vapor wall loss of glucose should be negligible when suspended particles exist. Oleic acid is an SVOC and so should have appreciable vapor wall losses over an aerosol suspension. shows the results of experiments with (a) glucose and (b) oleic acid.
Figure 1. Mean particle volume and org:sulf of ammonium sulfate particles containing glucose (a) and oleic acid (b). Symbols represent the change of organics on the ammonium sulfate seeds. Glucose (a) did not show any obvious change. Oleic acid (b) showed substantial vapor loss.
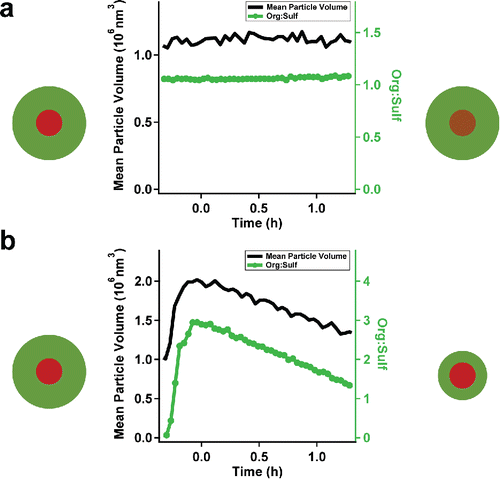
We prepared particles containing a mixture of ammonium sulfate and glucose by nebulizing an aqueous solution of glucose and ammonium sulfate. In all of the figures, t = 0 corresponds to the time we consider to be the start of “passive decay,” typically 5 min after injection (the mixing time of our chamber). Over 1.5 h we did not observe any significant change in the mean particle volume (obtained from the SMPS), or org:sulf (obtained from the AMS). This is consistent with glucose having no or very little vapor wall loss.
In contrast to glucose, oleic acid showed a clear decrease over time in both the mean particle volume and org:sulf, as shown in . We injected oleic acid with the vaporizer between −0.3 and −0.05 h, in the presence of ammonium-sulfate seeds. The results clearly indicate that oleic acid evaporated continuously from the particles. The particles lost half of their oleic acid in around 1 h. Considering that oleic acid vapors must already have reached their saturation concentration (4.0 µg m−3, May et al. Citation2012) before condensing onto the ammonium-sulfate seeds, the most probable, and perhaps only, sink for the lost oleic acid was the Teflon chamber walls. We further confirmed this by repeating the oleic-acid injection after the seed particles were nearly denuded. We again observed no nucleation as the seeds were re-coated, which was followed by oleic-acid loss at the earlier rate. The results clearly confirm that the vapor losses of SVOCs are significant and observable.
Mass balance of oleic acid during the injection
To quantify the loss rate, we need to determine the evaporation rate from the entire suspension accurately. To do this, we rely on multiple constraints from the SMPS and the AMS, but we shall start with a mass balance including the quantity of injected oleic acid as well. In , we compare the total injected amount of oleic acid with the amount of oleic acid we observed on the particles for a pair of injections. The trace on the top shows the SMPS-based total suspended particle volume concentration of bare ammonium-sulfate seeds that we then coated with oleic acid. This rises due to injection and falls due to wall losses (of both particles and vapors). We stopped seed injection at t = 0 h and then injected two aliquots of oleic acid in increments of 18 µg m−3 (at 100% injection efficiency, considering the chamber volume as 10 m3) at 0.8 h and 2.4 h. Each injection is marked by a vertical black bar extending upward from the particle volume at the injection time. The bars have two hash marks. The lower hash mark shows the particle volume extrapolated back to the injection time, based on an exponential fit (shown in cyan) to the subsequent volume measurements. The upper hash mark shows the estimated total injected concentration. The two measures disagree after the first injection by an amount comparable to the oleic-acid saturation concentration, 4.0 µg m−3. The two measures agree well after the second injection. We also measured org:sulf using the AMS, which allows us to constrain the overall particle wall loss; we show two points at the end of each evaporation period as squares in , along with an exponential fit (the trace on the bottom) to the sulfate mass concentration. This allows us to determine the absolute condensed-phase oleic-acid concentration, which we show on the right-hand y-axis in , from which we calculate the absolute evaporation rate.
Figure 2. Total volume concentration of ammonium sulfate particles as they are coated with oleic acid determined from SMPS measurements. Oleic acid injections in increments of 18 µg m−3 occurred at 0.8 h and 2.4 h. The squares are calculated sulfate concentrations based on the organic to sulfate mass ratio measured with an AMS. The curve shows steady exponential decay of the sulfate mass. The upper hash marks above the vertical lines show the injected oleic acid concentration. The curves are fits of the total volume concentration following each injection, and the lower hash marks are the extrapolation to the injection time. The gaps above the lines show the difference between the injected amount and the concentration on the particles. The right axis is scaled to the oleic acid mass concentration. The difference in the first injection is comparable to the oleic acid saturation concentration. The difference in the second injection was very small because the oleic acid in the gas phase was already saturated.
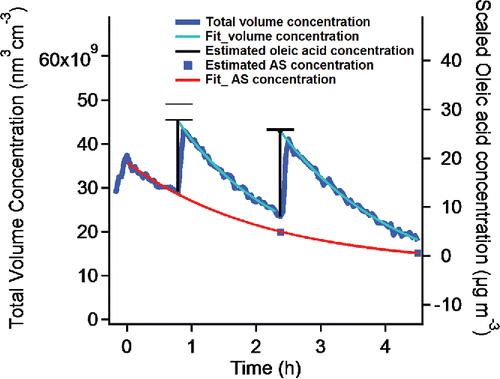
We conducted three such pairs of injections and summarize the results in . For each of the initial injections, the difference between the injected amount and the amount observed on the particles is comparable to the oleic-acid saturation concentration. For each of the subsequent injections, the differences are small. Our interpretation is that the initial oleic-acid injections first saturated the gas phase with vapors and then forced condensation to the ammonium-sulfate seeds, and that the subsequent injections into an already saturated system were exactly balanced by condensation to the seeds.
Table 2. The difference between the injected amounts and the estimated increases of the concentration of oleic acid on the particles in six injections.
The effect of size-dependent particle composition
Another possible complication is size-dependent particle wall loss. This could change the (bulk) org:sulf even without vapor losses when the particle composition is size dependent (which it is after condensation to seeds). In , we compare the behavior of particles that we formed via condensation of vaporized hexacosane to ammonium-sulfate seeds to the behavior of particles containing glucose and ammonium sulfate that we formed via atomization of a glucose and ammonium-sulfate solution. Both organics have very low vapor pressure. shows the mass distributions for the two systems. Hexacosane (top) condenses onto the ammonium-sulfate seeds and so is relatively more abundant on the smaller particles, which have a higher surface area to volume ratio (Donahue et al. Citation2014). Glucose (bottom) is part of a homogenous (dried) solution and so the composition is independent of size.
Figure 3. (a) The mass distribution of organic and sulfate of hexacosane and glucose coated ammonium sulfate particles. (b) The change of org:sulf of particles over the time. Mixed glucose and ammonium sulfate particles have a constant composition vs size. Hexacosane condensed onto ammonium sulfate leads to a larger organic mass fraction on smaller particles. However, org:sulf did not show any significant evolution for either particle type. Size dependent particle wall losses thus do not substantially complicate data interpretation.
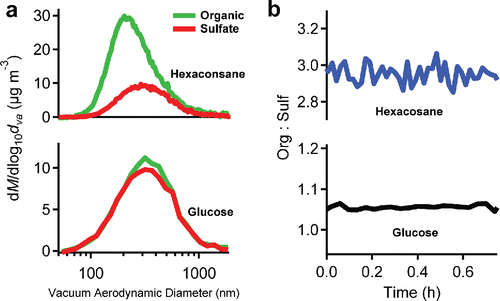
In , we show the measured org:sulf for both of these systems over time. Both are nearly constant, changing by much less than 10% h−1. Though one might expect smaller particles with higher org:sulf to be preferentially lost in the hexacosane system, if anything org:sulf in this case rises slightly during the experiment.
Wall loss of oleic acid vapor at the different temperatures
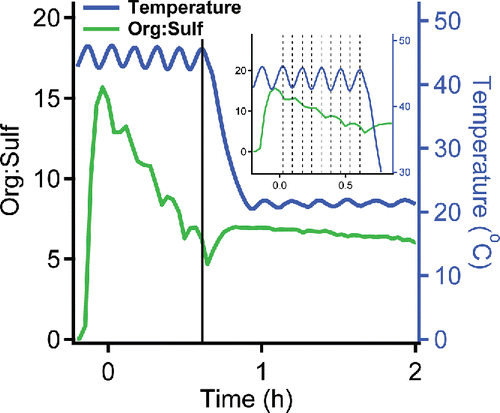
We also studied the vapor wall loss of oleic acid at different temperatures. The saturation concentrations of SVOCs increase sharply with temperature (Epstein et al. Citation2010). In , we show the evolution of org:sulf in a chamber initially held at 44°C, but subsequently cooled to 22°C. The org:sulf decreased rapidly at 44°C and much more slowly when we dropped the temperature to 22°C. The different depletion rate of org:sulf clearly illustrates that the vapor wall-loss rate of oleic acid is positively correlated with its concentration in the gas phase. Even the oscillation of org:sulf evident in the data is in phase with the (over-damped) temperature signal, indicating that the oleic-acid vapors equilibrated quickly with the particles (as expected). Finally, we observed a significant organic mass increase on the particles due to oleic-acid vapor condensation as the chamber cooled from 44°C to 22°C. These results confirm that the aerosol systems equilibrate quickly (Saleh et al. Citation2013), and that SVOCs with higher vapor concentrations show higher vapor wall-loss rates.
Figure 4. The change of org:sulf of ammonium sulfate particles coated with oleic acid inside the Teflon chamber at different temperatures. The loss rate of oleic acid dropped dramatically after the temperature changed from 44°C to 22°C. The insert shows that the oscillation of org:sulf was in phase with the (over-damped) chamber temperature.
Quantification of the vapor wall-loss rate
To quantify the vapor wall-loss rate, we must synthesize several measurements. As we show in , we consider the mass balance and dynamics of organics and sulfate (seeds) between the gas phase, the particle phase, and the Teflon chamber walls. We describe the net flux of organics to the gas phase from particles (, µg m−3 min−1) and to the walls (
, µg m−3 min−1) in Equations (Equation2
[2] ) and (Equation3
[3] ).
[2]
[3]
Scheme 2. The mass balance and dynamics of organics and sulfate (seeds) including the gas phase, the particle phase and the chamber walls. The ammonium sulfate constrains particle wall losses. The vapor wall loss rate of organics can be determined by the change of the organic to sulfate ratio in the particles (org:sulf) and the corrected suspended sulfate concentrations.
In these equations, (m min−1) is the mean thermal speed of the gas-phase molecules perpendicular to the particles,
is the effective mass accommodation coefficient (we assume
; Julin et al. Citation2014)
(m2) is the Fuchs-corrected surface area concentration of the suspended particles (accounting for gas-phase diffusion limitations in the transition regime; Trump et al. Citation2014). The suspended condensation sink is the product of these first three terms:
(min−1). The term in parentheses in Equation (Equation2
[2] ) is the thermodynamic driving force for evaporation. Within that,
(µg m−3) is the concentration of the organic in the gas phase,
(µg m−3) is the saturation concentration of the organic in the gas phase, and
is the activity of the organic in the particles. For pure organics,
.
To calculate the wall flux, we need to know (min−1), which is the diffusion corrected surface-area concentration (equivalent to the Fuchs corrected surface area) of the Teflon chamber walls (McMurry and Grosjean Citation1985). Here
is the activity of the organic at the wall. As we have shown for oleic acid, even after recharging the chamber with vapors, we observe almost complete evaporation from coated ammonium-sulfate particles, and so we shall treat vapor loss of SVOCs to the chamber walls as quasi-irreversible. For this reason, we set
.
Under almost all circumstances, the fluxes in Equations (Equation2[2] ) and (Equation3
[3] ) will balance and the organic vapor will be in a steady state. Thus,
=
and
[4]
is thus positively correlated with
and influenced by
and
.
The key parameters in these calculations are the condensation sinks of the particles and the Teflon chamber walls, and
. We calculate
using Equation (Equation5
[5] ) (Trump et al. Citation2014):
[5]
During our experiments, min−1.
Based on direct measurement of sulfuric-acid vapor loss using a nitrate-ion CIMS, we find min−1 (Hofbauer et al., in preparation), which is consistent with theoretical expectations for a turbulently mixed chamber, where
(McMurry and Grosjean Citation1985). We expect that gas-phase species with MW ∼ 150–300 g mol−1 should have loss rates within 20% of each other. To simplify our calculations, here we use the
value from sulfuric acid directly and assume that it remains constant for all experiments.
In our experiments, is 10–20 times larger than
, this means that the organic coated aerosols will sustain SVOCs in the gas phase within 10% of their saturation concentration throughout the experiments. We can thus simplify Equation (Equation5
[5] ) to obtain
[6]
For pure compounds, the equilibrium concentration of the organic () is approximately equal to the saturation concentration (
). The loss rate of organic vapors to the walls (
, µg m−3 min−1) is equal to the net evaporation rate of organics from the particles (
). In addition, it depends linearly on the saturation concentration (
), the condensed-phase activity (
and the condensation sink to the chamber walls (
).
To actually measure evaporation, we need to observe particles shrink, and the most precise method is to track org:sulf with the HR-ToF-AMS. However, we still need to relate this measurement to the absolute flux of organics from particles to the gas phase. As shown in , the decrease in the ammonium-sulfate mass concentration is only due to the loss of particles to the chamber walls (Equation (Equation7[7] )). In contrast, the decrease of organic signal includes two contributions. One is the loss of particles, and the other is the direct loss of organic vapor to the Teflon chamber walls, which is approximately equal to the organic mass loss from the particles (
) (Equation (Equation8
[8] )):
[7]
[8] where
(µg m−3) and
(µg m−3) are the organic and sulfate concentration in the particles, respectively, and
(min−1) is the particle wall-loss rate constant. Combining Equations (Equation7
[7] ) and (Equation8
[8] ), we obtain
[9]
[10]
Because solid and semi-solid particles bounce off of the AMS vaporizer, we must correct for particle collection efficiency (CE (ammonium sulfate) ∼ 0.2; Cross et al. Citation2009). Furthermore, CE varies with org:sulf. Fortunately, we can measure CE via LSSP as described above, obtaining CE(t) from a linear fit to the LSSP CE data. Accounting for CE, we obtain[11] where
and
are the raw sulfate and organic signals from the AMS measurement, respectively. The loss rate of organic vapor to the chamber walls (
) is proportional to the rate of change of the measured org:sulf but also the sulfate concentration at that time. We now define
(µg m−3) as the integrated mass loss of organic to the Teflon chamber walls over the time t:
[12]
We can calculate the total mass loss of organics, , adding the change of org:sulf over each measurement interval, multiplied by the sulfate concentration during each interval (1 min in this study, as V mode measurements from the HR-AMS are 1 min averages). After plotting the total mass loss with respect to time, we find the loss rate from the slope of a linear fit to those data.
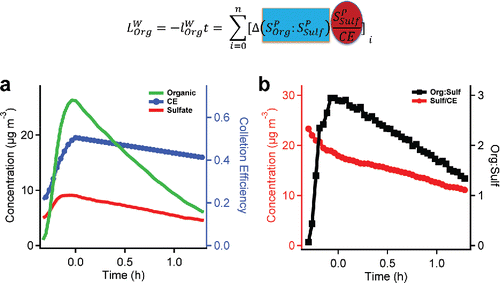
In , we illustrate the elements of this calculation. In , we show the concentrations of organics and sulfate measured with the HR-AMS during an experiment, along with the measured CE using the LSSP mode; CE rises from 0.2 to 0.5 as the organics coat the ammonium-sulfate seeds and then falls steadily as the coating evaporates. In , we show the resulting org:sulf and the CE-corrected sulfate concentration. These are the necessary terms for the integrated loss calculation, which we highlight with the respective colors in the embedded equation.
Figure 5. A graphical illustration of our method to quantitatively measure evaporation rates for a representative experiment. (a) The organic and sulfate concentration and the collection efficiency measured with LS-HR-AMS. (b) The org:sulf and the corrected sulfate concentration vs time (matching the highlights in the equation), which were calculated based on (a).
Calculation of the vapor wall-loss rates of oleic acid and levoglucosan
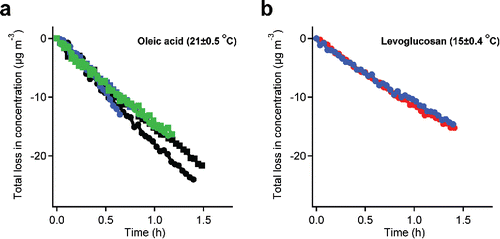
To test our ability to measure wall losses quantitatively, we measured evaporation from two systems that in theory should have similar saturation concentrations: oleic acid (at 21°C) and levoglucosan (at 15°C). In , we show the total evaporation loss over time that we obtain from Equation (Equation11[11] ). The results show very good reproducibility. Six measurements of oleic acid and two measurements of levoglucosan match each other very well. The good linear correlation shows that the loss of these vapors to the Teflon chamber walls follows near zeroth-order kinetics as expected, and that the vapor wall-loss rate was constant. The constant vapor wall-loss rate also indicates that the chamber walls were far from saturated. We determined the loss rates with a linear fit to find the slopes, which were 15.8 ± 0.1 µg m−3 h−1 for oleic acid, and 11.4 ± 0.1 µg m−3 h−1 for levoglucosan. We estimate the saturation concentrations of oleic acid to be 4.0 µg m−3 at 21°C and levoglucosan to be 2.9 µg m−3 at 15°C (May et al. Citation2012). This shows that SVOCs with similar saturation concentrations have similar vapor wall-loss rates.
Figure 6. The total vapor loss of oleic acid (a) and levoglucosan (b) over the time. The good linear correlation shows that the loss of oleic acid and levoglucosan vapor to the Teflon chamber walls follows near zeroth order kinetics as expected, and that the vapor wall loss rates are constant.
We also calculated the condensation sink to the chamber wall () using Equation (Equation6
[6] ). The values from experiments with oleic acid and levoglucosan are almost identical, ∼ 0.065 min−1. This is about half of the value measured directly for sulfuric acid vapor using a CIMS. However, we did not conduct the CIMS measurements concurrently with these observations but rather during a period when that instrument was not in use on a different experimental apparatus. Given the separation in time and the completely different experimental approaches, we regard these independent loss measurements as good agreement. The condensation sink to the particles (
min−1) for the SVOCs is 15–30 times larger than
. This confirms that the mass exchange of SVOCs between particles and vapors is much faster than the wall loss of vapors, and that the SVOC coated particles thus sustained SVOC vapors nearly at their saturation concentrations.
The wall loss of oleic acid vapors over organic solutions
To further investigate the relationship between the vapor wall-loss rate and the concentration of SVOC vapors in gas phase, we studied the vapor wall loss of oleic acid from oleic acid/d62-squalane mixtures. Following Raoult's law, the concentration of oleic-acid vapors in the gas phase should be proportional to its mole fraction in the mixture, or more correctly its activity, . This is also important broadly, as organics in atmospheric particles (and most chamber experiments) are almost always present in complex mixtures. Consequently, we measured the loss rate of oleic acid from a series of oleic acid/d62-squalane mixtures with different mole fractions of oleic acid. For this experiment, we considered d62-squalane to be nearly non-volatile.
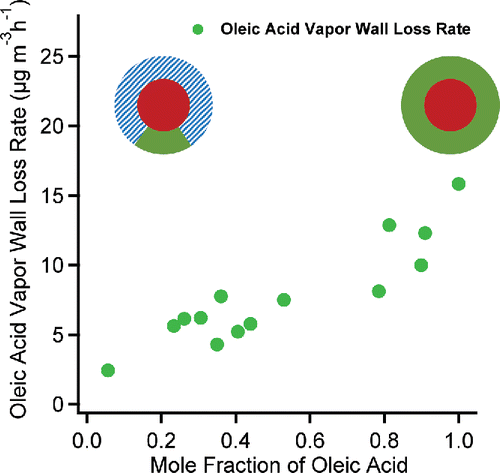
We used the fragment C4D9+ (m/z = 66) to calculate the concentration of d62-squalane. The signal ratio of fragment C4D9+ to d62-squalane was f66 = 0.243 for pure d62-squalane particles. We used the fragments, C4H7+ and C3H3O+ (m/z = 55) to calculate the concentration of oleic acid. The signal ratio of both fragments (C4H7++ C3H3O+) to oleic acid was f55 = 0.197 for pure oleic acid particles. In , we show the overall wall-loss rate of oleic-acid vapor as a function of the oleic-acid mole fraction of each mixture. As expected, we observed a higher loss rate of oleic acid when the mixture had a higher mole fraction of oleic acid. Pure oleic acid showed a larger loss rate than any mixture, and the rate was generally proportional to . This again demonstrates that the vapor wall-loss rates are proportional to the concentration of SVOC vapors in our Teflon chamber.
Figure 7. The loss rates of oleic acid in oleic acid/d62-squalane mixtures vs. the oleic acid mole fraction. The cartoon illustrates the mass fraction of oleic acid in the mixture of oleic acid /d62-squalane (dash). The loss rate increases with increasing oleic acid mole fraction in the mixture, which corresponds to a higher oleic acid vapor concentration in the gas phase.
Observation of the vapor wall loss of n-alkanes
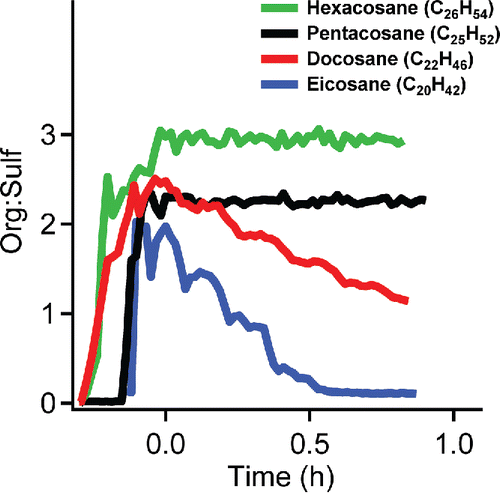
We also studied the vapor wall-loss behavior of four alkanes: hexacosane, pentacosane, docosane, and eicosane. In , we show the change of org:sulf for ammonium-sulfate particles coated by the alkanes. In each case, the passive decay starts at t = 0 h. Hexacosane and pentacosane show very small decreases in org:sulf. Both docosane and eicosane show steady decreases in org:sulf indicating continuous vapor loss to the Teflon chamber walls. The org:sulf of eicosane decreases faster than docosane, and reaches a value near 0 well before the end of the experiment, showing that all of the eicosane evaporates from the particles. Even for the most volatile species, the SVOC vapor wall loss appears to be nearly irreversible. The results clearly show that the vapor wall loss of the alkanes increases with decreasing carbon number and thus increasing saturation concentration.
Figure 8. The organic to sulfate ratio (org:sulf) vs time of ammonium sulfate particles coated by hexacosane, pentacosane, docosane, and eicosane. The passive decay started at time 0 h. The loss of the alkanes increases with decreasing carbon number and increasing saturation concentration. Eicosane shows the largest loss rate and almost disappears from particles after half an hour.
Summary of the vapor wall loss of all SVOCs in this study
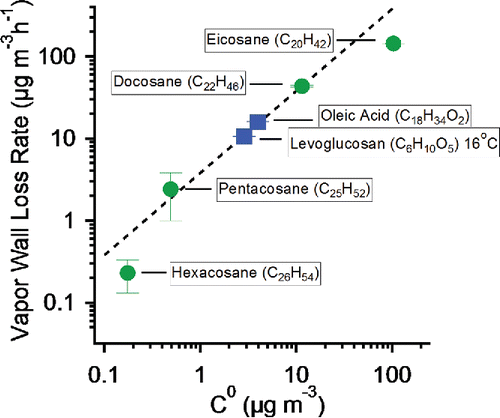
In , we show the measured vapor wall-loss rates for all of the SVOCs in this study vs. their saturation concentrations (Chickos and Hanshaw Citation2003; May et al. Citation2012) on a log-log plot. The wall-loss rate of SVOC vapors is clearly significant and proportional to the vapor concentration. The data are consistent with a line of slope 1 (a first-order dependence) at 3.8 h−1 (0.063 min−1). This is consistent with nearly irreversible vapor wall loss of SVOCs under typical chamber SOA conditions, with single, chamber-dependent wall condensation sink. This is consistent with the findings of Matsunaga and Ziemann, who suggest an equivalent organic concentration for Teflon walls for partitioning calculations of 2000–10,000 µg m−3, depending on molecular structure. Thus, even the most volatile SVOC we study here, eicosane, with C* = 100 µg m−3, would have an equilibrium vapor pressure over the Teflon walls of only about 1 µg m−3 after the walls absorbed a “saturation equivalent” of 100 µg m−3. Quasi-irreversible wall-loss rate-limited by diffusion through the boundary layer near the walls would greatly simplify wall loss corrections.
Figure 9. The loss rates of hexacosane, pentacosane, docosane, eicosane, levoglucosan and oleic acid, plotted vs saturation concentration. Alkanes have circle symbols. The vapor wall loss rates of SVOCs are significant and proportional to their vapor concentrations (C°). The black dashed line is a first-order fit with a wall condensation sink of 3.8 h−1.
We can also address the question of whether organic vapor accommodation to the suspended particles is small, as suggested in a recent paper (Zhang et al. Citation2014). Our results are consistent with the vapor-particle partitioning establishing a steady state far more rapidly than the vapor-wall exchange timescale, consistent with :
> 10. The excellent mass balance between injected oleic acid and the oleic acid we observed on the particles (and inferred in the gas phase) demonstrates that most oleic acid condensed on the particles during the injection. Another example is the rapid transition shown in from an increasing org:sulf while we were vaporizing oleic acid into the chamber to a much slower decrease in org:sulf due to vapor wall loss. These data are consistent with other findings showing accommodation coefficient αorg > 0.1 (Saleh and Khlystov Citation2009; Saleh et al. Citation2013; Julin et al. Citation2014; Palm et al. Citation2016). It is more difficult to assess the accommodation coefficient of organics to the Teflon wall, as vapor loss is rate-limited by diffusion for αwall > 10−4 or so (McMurry and Grosjean Citation1985). However, our results are consistent with αwall greater than this critical value. Thus, our results are consistent with reasonably rapid accommodation of vapors to the organic phase of suspended particles and sufficiently rapid accommodation of vapors to Teflon for gas-phase diffusion through the boundary layer to be rate limiting. Our results are inconsistent with slow accommodation in either case.
Conclusions
We have successfully developed a method to constrain the vapor wall loss of SVOCs due to the adsorption and/or absorption of organic vapor to the Teflon chamber walls with an HR-ToF-AMS. The parameter org:sulf measured for SVOC coated particles can be used to determine precisely if the vapor wall loss of SVOCs is significant. The vapor wall-loss rate of the SVOCs is proportional to both any decrease of org:sulf and the sulfate concentration, which can be calculated from the HR-ToF-AMS measurement. Our results clearly demonstrate that the wall-loss rate of SVOC vapors is significant and consistently proportional to the vapor concentration in the gas phase. Our results confirm that the wall loss of organic vapors needs to be considered in smog-chamber studies, especially when evaluating the SOA formation as well as OA evolution.
Funding
This research was supported by grant CHE1412309 from the National Science Foundation. The High-Resolution Aerosol Mass Spectrometer was purchased with Major Research Instrumentation funds from NSF CBET0922643 and the Wallace Research Foundation.
Related Research Data
References
- Chickos, J. S., and Hanshaw, W. (2003). Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from C21 to C30 at T = 298.15 K by Correlation Gas Chromatography. J. Chem. Eng. Data, 49:77–85.
- Chirico, R., DeCarlo, P. F., Heringa, M. F., Tritscher, T., Richter, R., Prévôt, A. S. H., Dommen, J., Weingartner, E., Wehrle, G., Gysel, M., Laborde, M., and Baltensperger, U. (2010). Impact of Aftertreatment Devices on Primary Emissions and Secondary Organic Aerosol Formation Potential from in-use Diesel Vehicles: Results from Smog Chamber Experiments. Atmos. Chem. Phys., 10:11545–11563.
- Cross, E. S., Onasch, T. B., Canagaratna, M., Jayne, J. T., Kimmel, J., Yu, X. Y., Alexander, M. L., Worsnop, D. R., and Davidovits, P. (2009). Single Particle Characterization Using a Light Scattering Module Coupled to a Time-of-flight Aerosol Mass Spectrometer. Atmos. Chem. Phys., 9:7769–7793.
- Donahue, N., Robinson, A., Trump, E., Riipinen, I., and Kroll, J. (2014). Volatility and Aging of Atmospheric Organic Aerosol, in Atmospheric and Aerosol Chemistry, V. F. McNeill and P. A. Ariya, eds., Springer, Berlin, Heidelberg, pp. 97–143.
- Donahue, N. M., Epstein, S. A., Pandis, S. N., and Robinson, A. L. (2011). A Two-dimensional Volatility Basis Set: 1. Organic-aerosol Mixing Thermodynamics. Atmos. Chem. Phys., 11:3303–3318.
- Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N. (2006). Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics. Environ. Sci. Technol., 40:2635–2643.
- Drewnick, F., Hings, S. S., DeCarlo, P., Jayne, J. T., Gonin, M., Fuhrer, K., Weimer, S., Jimenez, J. L., Demerjian, K. L., Borrmann, S., and Worsnop, D. R. (2005). A New Time-of-Flight Aerosol Mass Spectrometer (TOF-AMS)—Instrument Description and First Field Deployment. Aerosol Sci. Technol., 39:637–658.
- Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petaja, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F. (2014). A Large Source of Low-Volatility Secondary Organic Aerosol. Nature, 506:476–479.
- Epstein, S. A., Riipinen, I., and Donahue, N. M. (2010). A Semiempirical Correlation Between Enthalpy of Vaporization and Saturation Concentration for Organic Aerosol. Environ. Sci. Technol., 44:743–748.
- Grieshop, A. P., Miracolo, M. A., Donahue, N. M., and Robinson, A. L. (2009). Constraining the Volatility Distribution and Gas-Particle Partitioning of Combustion Aerosols Using Isothermal Dilution and Thermodenuder Measurements. Environ. Sci. Technol., 43:4750–4756.
- Hildebrandt, L., Donahue, N. M., and Pandis, S. N. (2009). High Formation of Secondary Organic Aerosol from the Photo-oxidation of Toluene. Atmos. Chem. Phys., 9:2973–2986.
- Hoffmann, T., Odum, J. R., Bowman, F., Collins, D., Klockow, D., Flagan, R. C., and Seinfeld, J. H. (1997). Formation of Organic Aerosols from the Oxidation of Biogenic Hydrocarbons. J. Atmos. Chem., 26:189–222.
- Huffman, J. A., Jayne, J. T., Drewnick, F., Aiken, A. C., Onasch, T., Worsnop, D. R., and Jimenez, J. L. (2005). Design, Modeling, Optimization, and Experimental Tests of a Particle Beam Width Probe for the Aerodyne Aerosol Mass Spectrometer. Aerosol Sci. Technol., 39:1143–1163.
- Julin, J., Winkler, P. M., Donahue, N. M., Wagner, P. E., and Riipinen, I. (2014). Near-Unity Mass Accommodation Coefficient of Organic Molecules of Varying Structure. Environ. Sci. Technol., 48:12083–12089.
- Kalberer, M., Paulsen, D., Sax, M., Steinbacher, M., Dommen, J., Prevot, A. S. H., Fisseha, R., Weingartner, E., Frankevich, V., Zenobi, R., and Baltensperger, U. (2004). Identification of Polymers as Major Components of Atmospheric Organic Aerosols. Science, 303:1659–1662.
- Kamens, R., Jang, M., Chien, C.-J., and Leach, K. (1999). Aerosol Formation from the Reaction of α-Pinene and Ozone Using a Gas-Phase Kinetics-Aerosol Partitioning Model. Environ. Sci. Technol., 33:1430–1438.
- Kokkola, H., Yli-Pirilä, P., Vesterinen, M., Korhonen, H., Keskinen, H., Romakkaniemi, S., Hao, L., Kortelainen, A., Joutsensaari, J., Worsnop, D. R., Virtanen, A., and Lehtinen, K. E. J. (2014). The Role of Low Volatile Organics on Secondary Organic Aerosol Formation. Atmos. Chem. Phys., 14:1689–1700.
- Liu, S., Russell, L. M., Sueper, D. T., and Onasch, T. B. (2013). Organic Particle Types by Single-particle Measurements Using a Time-of-flight Aerosol Mass Spectrometer Coupled with a Light Scattering Module. Atmos. Meas. Tech., 6:187–197.
- Loza, C. L., Chan, A. W. H., Galloway, M. M., Keutsch, F. N., Flagan, R. C., and Seinfeld, J. H. (2010). Characterization of Vapor Wall Loss in Laboratory Chambers. Environ. Sci. Technol., 44:5074–5078.
- Matsunaga, A., and Ziemann, P. J. (2010). Gas-Wall Partitioning of Organic Compounds in a Teflon Film Chamber and Potential Effects on Reaction Product and Aerosol Yield Measurements. Aerosol Sci. Technol., 44:881–892.
- May, A. A., Saleh, R., Hennigan, C. J., Donahue, N. M., and Robinson, A. L. (2012). Volatility of Organic Molecular Markers Used for Source Apportionment Analysis: Measurements and Implications for Atmospheric Lifetime. Environ. Sci. Technol., 46:12435–12444.
- McMurry, P. H., and Grosjean, D. (1985). Gas and Aerosol Wall Losses in Teflon Film Smog Chambers. Environ. Sci. Technol., 19:1176–1182.
- McVay, R. C., Cappa, C. D., and Seinfeld, J. H. (2014). Vapor-Wall Deposition in Chambers: Theoretical Considerations. Environ. Sci. Technol., 48:10251–10258.
- Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H. (1996). Gas/particle Partitioning and Secondary Organic Aerosol Yields. Environ. Sci. Technol., 30:2580–2585.
- Odum, J. R., Jungkamp, T. P. W., Griffin, R. J., Flagan, R. C., and Seinfeld, J. H. (1997). The Atmospheric Aerosol-Forming Potential of Whole Gasoline Vapor. Science, 276:96–99.
- Palm, B. B., Campuzano-Jost, P., Ortega, A. M., Day, D. A., Kaser, L., Jud, W., Karl, T., Hansel, A., Hunter, J. F., Cross, E. S., Kroll, J. H., Peng, Z., Brune, W. H., and Jimenez, J. L. (2016). In Situ Secondary Organic Aerosol Formation from Ambient Pine Forest Air Using an Oxidation Flow Reactor. Atmos. Chem. Phys., 16:2943–2970.
- Pandis, S. N., Paulson, S. E., Seinfeld, J. H., and Flagan, R. C. (1991). Aerosol Formation in the Photooxidation of Isoprene and β-pinene. Atmos. Environ., 25A:997–1008.
- Pankow, J. F. (1994). An Absorption Model of Gas/Particle Partitioning of Organic Compounds in the Atmosphere. Atmos. Environ., 28:185–188.
- Presto, A. A., Huff Hartz, K. E., and Donahue, N. M. (2005a). Secondary Organic Aerosol Production from Terpene Ozonolysis. 2. Effect of NOx Concentration. Environ. Sci. Technol., 39:7046–7054.
- Presto, A. A., HuffHartz, K. E., and Donahue, N. M. (2005b). Secondary Organic Aerosol Production from Terpene Ozonolysis. 1. Effect of UV Radiation. Environ. Sci. Technol., 39:7036–7045.
- Robinson, A. L., Donahue, N. M., Shrivastava, M. K., Weitkamp, E. A., Sage, A. M., Grieshop, A. P., Lane, T. E., Pierce, J. R., and Pandis, S. N. (2007). Rethinking Organic Aerosols: Semivolatile Emissions and Photochemical Aging. Science, 315:1259–1262.
- Robinson, E. S., Saleh, R., and Donahue, N. M. (2013). Organic Aerosol Mixing Observed by Single-Particle Mass Spectrometry. J. Phys. Chem. A, 117:13935–13945.
- Robinson, E. S., Saleh, R., and Donahue, N. M. (2015). Probing the Evaporation Dynamics of Mixed SOA/Squalane Particles Using Size-Resolved Composition and Single-Particle Measurements. Environ. Sci. Technol., 49:9724–9732.
- Sage, A. M., Weitkamp, E. A., Robinson, A. L., and Donahue, N. M. (2008). Evolving Mass Spectra of the Oxidized Component of Organic Aerosol: Results from Aerosol Mass Spectrometer Analyses of Aged Diesel Emissions. Atmos. Chem. Phys., 8:1139–1152.
- Saleh, R., Donahue, N. M., and Robinson, A. L. (2013). Time Scales for Gas-Particle Partitioning Equilibration of Secondary Organic Aerosol Formed from Alpha-Pinene Ozonolysis. Environ. Sci. Technol., 47:5588–5594.
- Saleh, R., and Khlystov, A. (2009). Determination of Activity Coefficients of Semi-Volatile Organic Aerosols Using the Integrated Volume Method. Aerosol Sci. Technol., 43:838–846.
- Stanier, C. O., Donahue, N., and Pandis, S. N. (2008). Parameterization of Secondary Organic Aerosol Mass Fractions from Smog Chamber Data. Atmos. Environ., 42:2276–2299.
- Stern, J. E., Flagan, R. C., Grosjean, D., and Seinfeld, J. H. (1987). Aerosol Formation and Growth in Atmospheric Aromatic Hydrocarbon Photooxidation. Environ. Sci. Technol., 21:1224–1231.
- Trump, E. R., Riipinen, I., and Donahue, N. M. (2014). Interactions Between Atmospheric Ultrafine Particles and Secondary Organic Aerosol Mass: A Model Study. Boreal Environ. Res., 19(5):352–362.
- Weitkamp, E. A., Sage, A. M., Pierce, J. R., Donahue, N. M., and Robinson, A. L. (2007). Organic Aerosol Formation from Photochemical Oxidation of Diesel Exhaust in a Smog Chamber. Environ. Sci. Technol., 41:6969–6975.
- Yeh, G. K., and Ziemann, P. J. (2014). Alkyl Nitrate Formation from the Reactions of C8–C14 n-Alkanes with OH Radicals in the Presence of NOx: Measured Yields with Essential Corrections for Gas–Wall Partitioning. J. Phys. Chem. A, 118:8147–8157.
- Zhang, S.-H., Shaw, M., Seinfeld, J. H., and Flagan, R. C. (1992). Photochemical Aerosol Formation from α-Pinene and β-Pinene. J. Geophys. Res., 97(D18):20717–20729.
- Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H. (2014). Influence of Vapor Wall Loss in Laboratory Chambers on Yields of Secondary Organic Aerosol. Proc. Natl. Acad. Sci., 111:5802–5807.
- Zhang, X., Schwantes, R. H., McVay, R. C., Lignell, H., Coggon, M. M., Flagan, R. C., and Seinfeld, J. H. (2015). Vapor Wall Deposition in Teflon Chambers. Atmos. Chem. Phys., 15:4197–4214.