
ABSTRACT
Aerosol formation is directly influenced by meteorological properties such as temperature and relative humidity. This study examines the influence of temperature on the physical properties and chemical composition of the aerosol produced from radical oxidation of aliphatic amines. Aerosol formation for temperatures ranging from 10 to 40°C was investigated in dual 90 m3 indoor atmospheric chambers. Further, chemical and physical responses of aerosol formed at one temperature and then raised/cooled to another were investigated in detail. Around two to three times more aerosol formation occurred at 10°C than at 40°C. This has important implications for locations influenced by amine emissions during the winter months. Significant aerosol formation occurred with the oxidation of amines with nitrate radical (100–600 μg/m3) and consisted largely of amine nitrate salts. These reactions are important contributors to aerosol formation during the nighttime hours, when nitrate radical is the dominant oxidant and temperatures tend to be cooler. Solid/gas partitioning of amine nitrate salt aerosol was consistent with literature results. A novel, temperature dependent, mechanism describing peroxy and hydroperoxy radical reactions was observed in the trimethylamine with hydroxyl radical oxidation experiments.
Copyright © 2016 American Association for Aerosol Research
EDITOR:
1. Introduction
Secondary organic aerosol (SOA) and fine particulate matter have negative effects on human health (Dockery Citation2009; Pope et al. Citation1995), air quality, visibility (Watson Citation2002), and climate (IPCC Citation2013). Reduced nitrogen compounds, including ammonia and amines, have been shown to be significant precursors to aerosol formation in the atmosphere (Ge et al. Citation2011a). There are both anthropogenic and biogenic sources of amines, which include agricultural emissions (Mosier et al. Citation1973), by-products of selective catalytic reduction (Cadle and Mulawa Citation1980) and carbon capture control technologies (Borduas et al. Citation2013), and biomass burning (Westerholm et al. Citation1993; Ge et al. Citation2011a). Recent evidence has pointed to the growing importance of amines in new particle formation. There have been some studies looking at the formation of organic salt aerosol through the reactions of atmospherically relevant acids (HNO3, H2SO4) with alkyl amines (Barsanti et al. Citation2009). In addition to organic salt formation, field studies have observed that a significant portion of new particle growth is due to oxidized organic amine aerosol (Tan et al. Citation2002; Silva et al. Citation2008; Smith et al. Citation2008).
The atmospheric reactions of amines, however, are poorly understood. There have been a few previous atmospheric chamber studies investigating the oxidation of alkyl amines at room temperature (Malloy et al. Citation2009a, Lee and Wexler Citation2013; Price et al. Citation2014; Tang et al. Citation2014). These studies highlighted the presence and importance of salt formation, the formation of imines and carcinogenic nitramines, the formation of highly oxidized oligomers (trimethylamine experiments), and cloud condensation nuclei activity in secondary organic aerosol formation from alkyl amines. However, due to the wide range in volatility of alkyl amine salts (solid/gas equilibrium (Kp) from 1.65E-06 to 7.01E-09 at 25°C, (Ge et al. Citation2011b), the impact of a range of atmospherically relevant temperatures on SOA formation from alkyl amines is important for understanding the true impact and importance of these poorly understood aerosol precursors.
Temperature has a complicated impact on the gas-to-particle partitioning of secondary organic aerosol, as seen in the gas-to-particle partitioning equation (Odum et al. Citation1996):where R is the ideal gas constant, T is the ambient temperature, MW is the molecular weight,
is the activity coefficient, and
is the saturation liquid vapor pressure. It should be noted that temperature is not only seen in the numerator of the equation but exponentially in the denominator. Along with seasonal and regional temperature variations, there is significant temperature variation within the vertical temperature profile of the atmosphere. The effect of temperature becomes particularly complicated when applied to amine salts and semi-volatile organic amine compounds due to their vapor pressures being in a sensitive temperature range. These salts are formed in areas with high agricultural and combustion emissions (e.g., San Joaquin Valley, California; Logan, Utah; Toronto, Canada). To the authors' knowledge, the effect of ambient temperature on amine SOA formation has not been previously studied. Indeed, temperature effects on SOA formation from other compounds is limited and has focused on other biogenic and combustion emissions (Takekawa et al. Citation2003; Warren et al. Citation2009; Qi et al. Citation2010; Clark et al. Citation2016). This study examines the influence of temperature ranging from 10°C and 40°C on the physical properties and chemical composition of the aerosol produced from radical oxidation of aliphatic amines.
2. Methods
A set of well-characterized atmospheric chamber experiments were conducted on three aliphatic amines: trimethylamine (TMA, 25 wt.% solution in water, Sigma-Aldrich), diethylamine (DEA, >99.5%, Sigma-Aldrich), and butylamine (BA, 99.5%, Sigma-Aldrich). Tertiary, secondary, and primary aliphatic amines were chosen for this study to represent a range of emissions from agricultural facilities. All experiments were conducted in the UCR/CE-CERT dual 90 m3 atmospheric chambers. The chamber facility has been described in detail previously (Carter et al. Citation2005). The environmental chambers are housed in a 6 m × 6 m × 12 m thermally insulated enclosure and kept at a constant positive differential pressure of >0.0175” H2O. Photooxidation, when desired, was driven by a bank of 115W black lights with peak intensity of 350 nm (350 BL, Sylvania). The interior of the enclosure is lined with reflective anodized aluminum sheeting to ensure maximum intensity and even distribution of light. Prior to each experiment, the chambers were flushed with dry (RH < 0.001%) purified air (Aadco 737 air purification system) so that background particle concentration, NOx, and hydrocarbon concentrations were below the detection limits of the instruments used. There were no seed aerosols injected. Experiments were initiated at either 10°C or 40°C and then temperature was cycled between 10 and 40°C over the course of aging (). Amine concentrations were allowed to stabilize in the Teflon chambers before radical injection. Hydroxyl (OH) radicals (no NOx experiments) were produced from direct photolysis of hydrogen peroxide (H2O2) (Sigma Aldrich, 50wt% in H2O, stabilized). The nitrate radical (NO3) was formed from thermal decomposition of dinitrogenpentoxide (N2O5) (synthesized in-house by adding O3 to a flow of NO2 gas and collecting the resultant N2O5 in a −70°C cooling trap) (Griffin et al. Citation1999). Both oxidant precursors were injected into the chamber in a stream of heated nitrogen. OH experiments were designed to represent daytime chemistry while the NO3 experiments represented nighttime chemistry.
Table 1. Environmental chamber experiments.
A suite of instruments surround the chamber enclosure. The instruments are kept at room temperature (20°C) with insulated sampling lines leading from the chambers to reduce room temperature effects. The chemical composition of the aerosol products was measured by a High Resolution Time of Flight Aerosol Mass Spectrometer (HR-ToF-AMS; Aerodyne Research Inc.) (Jayne et al. Citation2000; DeCarlo et al. Citation2006). Briefly, an aerosol sample was drawn through a time-of-flight region where the particles were separated based on their vacuum aerodynamic diameter. The sample was vaporized by a 600°C oven followed by 70 eV electron impact ionization. The resulting ions pass through another time-of-flight section which can be operated in two flight path configurations, V and W. The shorter flight path (V-mode) provides better sensitivity at unit mass resolution. The longer flight path (W-mode) provides sufficient mass spectral resolution (4300 at m/z 200) to separate isobaric compounds and determine empirical formulas.
A custom built scanning mobility particle sizer (SMPS) (Cocker et al. Citation2001), was used to measure the concentration and size distribution (27–712 nm) of the aerosol formed during the experiments. The SMPS is housed inside the chamber enclosure in order to measure the aerosol properties at the current chamber temperature. The SMPS also communicates size distribution values in real-time to a pair of custom built instruments designed to measure particle density and volatility.
Particle density was measured by an aerosol particle mass analyzer (APM, Kanomax model 3600) in series with an SMPS. Details of the instrument are described previously (Ehara et al. Citation1996; Malloy et al. Citation2009b). Briefly, the mass of the particle is selected by the APM based on the peak number electrical mobility diameter identified by the independent SMPS. The mass selected particles are then measured by an in-series SMPS to obtain the peak number electrical mobility diameter transmitted by the APM. Density measurements were taken every 85 s.
Particle volatility was measured by a custom built volatility tandem differential mobility analyzer (VTDMA). The particles entering the first DMA column are size selected based on the peak number electrical mobility diameter provided by the independent SMPS. The resultant monodisperse aerosol is then sent through a Dekati® Thermodenuder (TD, 40°C, residence time: ∼17 s). The second DMA column scans the size distribution of the particles exiting the TD to determine the final diameter (Df). Particle volatility can then be expressed as the volume ratio before and after the TD or Volume Fraction Remaining [VFR, (Df/Di)3]. Volatility measurements were taken every 85 s.
3. Results and discussion
3.1. Aerosol formation
Particle formation (not wall loss corrected, WLC) along with temperature gradients for each of the experiments are shown in . The non-WLC mass concentration profiles display the changes due to temperature in each of the experiments and show the complete loss of particles after heating the chamber in the NO3 oxidation experiments. Therefore, only the non-WLC mass concentration profiles are shown in this study.
Figure 1. Mass concentration profiles (10 min averages) for the trimethylamine ( - circle), diethylamine ( - square), and butylamine (Δ - triangle) oxidation experiments (CHC = cold-hot-cold, HCH = hot-cold-hot).
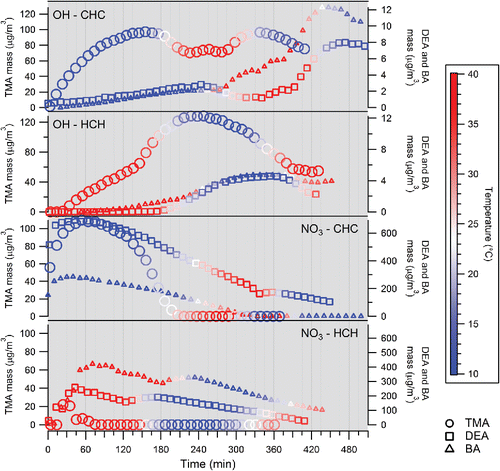
3.1.1. OH oxidation experiments
Aerosol formation was significantly greater starting at cold temperature (10°C) than at hot temperature (40°C) in each of the OH oxidation experiments. An interesting pattern is observed when the temperature is cycled. Mass concentration drops with increasing chamber temperature as the aerosol formed at cold temperature partitions to the gas phase. After cooling the chamber to the original temperature, those gas phase products partition back to the particle phase. This trend was also observed in the hot start experiments. This suggests that the reaction products of OH oxidation include semi-volatile organic amine compounds that partition to the particle phase at colder temperatures.
Greater aerosol formation occurred much faster in both TMA + OH oxidation experiments (97 μg/m3 cold and 65 μg/m3 hot within 150 min) than in the DEA and BA + OH experiments (5–12 μg/m3 after 450 minutes). This is likely due to the presence of a unique reaction pathway with TMA, as discussed in a previous study (Price et al. Citation2014), leading to the formation of highly oxidized oligomer compounds. The low volatility of these oligomers allows them to form readily at 40°C. The striking change in mass concentration (∼40 μg/m3) observed in both TMA + OH experiments indicates the significant formation of semi-volatile compounds that are present in the gas phase at 40°C and partition to the particle phase at 10°C. A possible reaction mechanism explaining the formation of these semi-volatile compounds is discussed in Section 3.4.4.
Similar aerosol formation trends were observed between the DEA and BA + OH experiments. The maximum aerosol formed at the end of the cold start DEA and BA + OH experiments is higher than would be expected through further oxidation of the precursor amine without temperature change. This indicates that a secondary formation event occurred in addition to condensational particle growth after cooling down the chamber to the original temperature. Unlike the DEA + OH oxidation experiments, particle formation was able to occur at 40°C in the BA + OH oxidation experiments.
3.1.2. NO3 oxidation experiments
A significant amount of aerosol (>100 μg/m3) was formed in the NO3 oxidation experiments (). The maximum aerosol concentration in the cold start TMA + NO3 experiment (110 μg/m3) was similar to that in the cold start TMA + OH experiment (97 μg/m3). The DEA and BA + NO3 experiments, on the other hand, produced far more aerosol than the DEA and BA + OH experiments (up to 86x more aerosol in the cold start experiments and up to 100x more aerosol in the hot start experiments). The overall aerosol formation was greatest in the cold start experiments.
A large particle formation event occurred in the cold start TMA + NO3 oxidation experiment (719,000 #/cm3 within 2 min of oxidation) compared to the cold start TMA + OH oxidation experiment (30,000 #/cm3 within 20 min of oxidation). The particles formed in the cold start TMA + NO3 experiment completely partition to the gas phase with increasing temperature. This is consistent with the formation of volatile amine nitrate salts through the following reactions:
Unlike the TMA + OH experiments, the particles lost to the gas phase at higher temperatures in the TMA + NO3 experiment do not return after cooling. This suggests that further oxidation of the gas phase amine products changes the gas to particle equilibrium (i.e., the amine precursor is completely oxidized and the oxidized amine products do not produce amine salts as readily as the precursor amine). Particles had difficulty forming at 40°C initial temperature for the NO3 oxidation experiment and quickly partitioned to the gas phase. There was no subsequent particle formation after cooling. Aerosol formation was greater at cold temperature (110 μg/m3) than at hot temperature (22 μg/m3) in the TMA + NO3 oxidation experiments.
Large particle formation occurred in the cold start DEA + NO3 oxidation experiment (). However, these particles, unlike the TMA + NO3 oxidation generated particles, do not completely partition to the gas phase with increasing temperature. A slight increase in mass concentration occurred after cooling the chamber back to the original temperature. This suggests the formation of an amine nitrate aerosol with a lower volatility than for the TMA + NO3 experiment. Diethylammonium nitrate salt (DEA·HNO3) formation could explain these observations. The solid/gas dissociation constant (Kp) for several amine salts at various temperatures were reported in Ge et al. (Citation2011b). The Kp for DEA·HNO3 was orders of magnitude lower than for TMA·HNO3 throughout the temperature range of −10°C to 50°C (e.g., 7.01E-09 and 1.65E-06, respectively at 25°C). In addition, Ge et al. (Citation2011b) found that Kp for amine salts increased by an order of magnitude for every 10°C increase in temperature. An equation to calculate Kp at different temperatures was obtained by fitting an exponential curve to the Kp values obtained by Ge et al. (Citation2011b). The temperature at which the aerosol begins to dissociate quickly in the cold start TMA + NO3 experiment is around 15°C. At that temperature, the calculated Kp for TMA·HNO3 is 2.0E-07 Pa2. For DEA·HNO3, a Kp value of 2.0E-07 Pa2 is reached at approximately 39°C, which is around the maximum temperature attained in the chamber. This could explain why the DEA·HNO3 aerosol formed in the cold start DEA + NO3 experiment is not greatly affected by the temperature range in this study. Less aerosol formation occurred at the beginning of the hot start DEA + NO3 oxidation experiment relative to the cold start experiment. Particle mass concentration increased due to condensational growth after cooling the chamber.
The overall trends in the BA + NO3 oxidation experiments () were similar to those observed in the TMA and DEA + NO3 oxidation experiments. Rapid particle growth occurred in the cold start NO3 oxidation experiment. These particles completely partition to the gas phase as temperature increased past 35°C, consistent with the formation of amine nitrate salts. The particles lost after heating the chamber do not return after cooling. About 30–50% less BA and oxidant were injected in the cold start BA + NO3 oxidation experiment due to an issue with the injection system. Therefore, more aerosol formation was observed in the hot start BA + NO3 experiment, and the aerosol does not completely partition to gas phase by the end of the experiment. Particle mass concentration increased due to condensational growth after cooling the chamber in the hot start BA + NO3 experiment.
3.2. Aerosol density
The average density value at the cold (10°C) and hot (40°C) temperatures for each amine oxidation experiment are provided in . The density generally decreases in the cold start TMA, DEA, and BA + OH experiments (1.78-1.41 g/cm3, 1.93-1.54 g/cm3, and 1.58-1.24 g/cm3, respectively). There is a sharp decrease in density from cold to hot and then a slight decrease from hot back to cold. This indicates that the higher density particles formed at cold temperatures partition to gas phase at warmer temperatures, but do not return to particle phase after cooling. This could be due to further gas phase oxidation forming lower density aerosol products. The hot start TMA + OH experiment density follows the same trend; however, the starting density is lower than in the cold start experiment (1.69–1.47 g/cm3). The instrument was offline at the beginning of the hot start DEA + OH experiment. However, both the SMPS and HR-ToF-AMS instruments indicate that particle formation did not occur until after the chamber was cooled. When particle formation did occur, the density value was not as high as in the cold start experiment and dropped over time (1.78–1.46 g/cm3). In the hot start BA + OH experiment, the density started out low (1.27 g/cm3), increased after cooling the chamber (1.42 g/cm3), and then gradually decreased over time (1.33 g/cm3). As in the hot start DEA + OH experiment, there was little particle formation at the beginning of the hot start BA + OH experiment. The increase in density after cooling could be due to the increase in particle formation. For the OH oxidation experiments, if the aerosol density were entirely dependent on temperature, the density would be expected to return to the original value after returning the chamber to the start temperature. Since the density generally decreases over time, it appears that further oxidation of the reaction products has a greater influence on density than changes in temperature alone.
Table 2. Average density, volatility, and elemental ratio (HR-ToF-AMS) values at 10°C and 40°C for each of the amine oxidation experiments.
Density values are not shown above 33°C for the TMA + NO3 oxidation experiments due to the loss of particles after heating. Overall, the density is lower in the DEA and BA + NO3 experiments than in the DEA and BA + OH experiments. There does not appear to be a significant change in density with temperature change in the DEA + NO3 experiments. However, the density is slightly lower for the hot start DEA + NO3 oxidation aerosol than for the cold start aerosol (1.17 vs. 1.32 g/cm3). The BA + NO3 experiments follow the same density trends as the DEA + NO3 experiments with lower density for the hot start BA + NO3 experiment than for the cold start BA + NO3 experiment (1.14 vs. 1.56 g/cm3).
3.3. Aerosol volatility
The average volume fraction remaining (VFR) at the cold (10°C) and hot (40°C) temperatures for each amine oxidation experiment are provided in . The thermodenuder (TD) was set to 40°C in experiments 1, 4, 5, 6, 9, and 12. The aerosol produced in each of the OH oxidation experiments increases in VFR over the course of the experiment, suggesting the formation of less volatile aerosol as the experiment progresses. As expected, the average VFR increases to near unity as the chamber temperature matches the thermodenuder temperature (40°C). The aerosol formed in both TMA + OH experiments was less volatile (VFR = 0.68-0.80) than that formed in the DEA + OH (VFR = 0.49-0.66) and BA + OH experiments (VFR = 0.65-0.69). In the hot start TMA + OH and BA + OH experiments (experiments 2 and 10), the TD was set to 100°C instead of 40°C, therefore producing a lower VFR than expected.
Highly volatile aerosol formed in each of the NO3 oxidation experiments. The TD was also set to 100°C in experiments 3, 7, 8, and 11. The particles completely partitioned to the gas phase after heating to 100°C in the TD, consistent with observed particle loss at chamber temperatures of 40°C for the TMA and BA experiments and indicative of amine salt aerosol. There appears to be no change in volatility with changing temperature in each of the NO3 oxidation experiments with the exception of the hot start BA experiment (exp. 12). Volatile aerosol was produced in the hot start BA + NO3 experiment (VFR = 0.30), with volatility gradually decreasing over the course of the experiment.
3.4. Aerosol chemical composition: Mass spectra
The HR-ToF-AMS mass spectra for the cold start experiments are presented in Figure S1 in the online supplemental information. The hot start experiment mass spectra are shown in Figure S2. The mass spectra for each of the experiments are shown in logarithmic scale to highlight the higher m/z peaks, i.e., m/z peaks greater than that of the amine precursor (TMA = 59 amu, DEA = 73 amu, BA = 73 amu).
3.4.1. TMA mass spectra
The mass spectra for both the cold and hot start TMA + OH experiments (Figures S1a–c and S2a–c) contain higher m/z peaks at 73 (C2H3NO2+), 88 (C3H6NO2+), 104 (C3H6NO3+), 145 (C5H9N2O3+), 161 (C5H9N2O4+), and 191 (C6H11N2O5+), which are fragment ions indicative of oligomer compounds. The mechanism describing the formation of these oligomer compounds is discussed in detail in Price et al. (Citation2014). Briefly, oligomers are produced through peroxy radical chemistry (RO2· + RO2·) that creates a peroxide linkage (R-O-O-R). The lower m/z peaks (<59) at 29 (CHO+), 30 (CH2O+, CH4N+), 44 (CO2+, CH2NO+), 45 (CH3NO+), and 58 (C2H4NO+, C3H8N+) are smaller aldehyde and amide fragment ions also expected from the oligomer compounds.
In contrast to the OH experiments, the mass spectra for the cold and hot start TMA + NO3 experiments (Figures S1d–f and S2d–f) are dominated by lower m/z peaks (<59) at 29 (CHO+), 30 (CH4N+), 42 (C2H4N+), 44 (C2H6N+), and 58 (C3H8N+), which are fragments of the unoxidized parent amine. This is consistent with the fragmentation of trimethylammonium nitrate salt (TMA·HNO3). The significant nitrate peaks at m/z 30 (NO+) and 46 (NO2+) also point to TMA·HNO3 formation. The higher m/z peaks (>59) at 73 (C4H11N+), 86 (C5H12N+), 100 (C5H8O2+), 119 (C6HNO2+), 133 (C4H7NO4+), 147 (C5H9NO4+), and 169 (C5HN2O5+) indicate that some additional oligomer chemistry is occurring. However, these oligomer fragments are much lower in concentration than the TMA·HNO3 fragments. Both the salt and oligomers are volatile based on loss at 40°C.
3.4.2. DEA mass spectra
The mass spectra for the cold and hot start DEA + OH experiments (Figures S1g–i and S2g–i) show lower m/z peaks (<m/z of the precursor amine, DEA = 73 amu) at 30 (CH4N+, CH2O+), 44 (CO2+, C2H6N+), 58 (C3H8N+, C3H6O+), and 73 (C3H5O2+, C4H11N+). These peaks are a mixture of fragments from amine and oxidized hydrocarbon aerosol. The lower m/z peaks (<73) at 15 (CH3+), 29 (CHO+, C2H5+), 43 (C2H3O+, C3H7+), 55 (C3H3O+, C4H7+), and 69 (C4H5O+, C5H9+) are fragments of hydrocarbon aerosol. The higher m/z peak (>73) at 86 (C4H8NO+) is the fragment of an oxidized parent amine molecule. The C3–C5 hydrocarbon fragments suggest that some type of addition chemistry is occurring, as C2 is the largest hydrocarbon chain on DEA. Amine type peaks (e.g., m/z 58) dominate during cold temperatures (10°C) while hydrocarbon peaks (e.g., m/z 55 and 69) are the more significant peaks at the hot temperatures (40°C). Both amine and hydrocarbon peaks are observed after the chamber returns to the original temperature.
Most of the m/z fragments from the vaporized (600°C) and ionized aerosol for the cold and hot start DEA + NO3 experiments (Figures S1j–l and S2j–l) were in the lower m/z range (≤73) at 15 (CH3+), 30 (CH4N+), 44 (C2H6N+), 58 (C3H8N+), and 73 (C4H11N+). These peaks are fragments of the unoxidized parent amine, consistent with the fragmentation of diethylammonium nitrate salt (DEA·HNO3). The significant nitrate peaks at m/z 30 (NO+) and 46 (NO2+) also support DEA·HNO3 formation. Although not as striking as in the TMA + OH experiments, there are higher m/z peaks (>73) at 146 (C4H6N2O4+) and 170 (C6H6N2O4+) that suggest some addition chemistry is occurring, possibly forming dimers. There was little change in chemical composition with temperature, consistent with the observations for DEA + NO3 aerosol density and volatility.
3.4.3 BA mass spectra
The mass spectra for the cold and hot start BA + OH experiments (Figures S1m–o and S2m–o) show lower m/z peaks (<m/z of the precursor amine, BA = 73 amu) at 30 (CH4N+, CH2O+), 44 (CO2+, C2H6N+), 58 (C3H8N+, C3H6O+), and 72 (C4H10N+, C4H8O+), which are a mixture of fragments from amine and oxidized hydrocarbon aerosol. The lower m/z peaks (<73) at 15 (CH3+), 29 (CHO+, C2H5+), 41 (C3H5+), 43 (C2H3O+, C3H7+), and 55 (C3H3O+, C4H7+) are fragments of hydrocarbon aerosol. All of the lower mass peaks can be explained through fragmentation of the oxidized products of the precursor amine. A series of higher m/z peaks (>73) at 142 (C8H16NO+), 158 (C8H16NO2+), and 174 (C8H16NO3+) grew in over the course of the cold and hot start BA + OH experiments. These higher m/z peaks were observed in a previous study (BA + O3/NO; Malloy et al. Citation2009a) and suggest dimerization of the oxidized amine products, perhaps through aldol addition/condensation reactions. The concentrations of these peaks increase at higher chamber temperatures. This indicates that the oxidized dimers form in the gas phase prior to particle phase partitioning while the dimer precursors are able to partition to the particle phase at the colder temperatures.
The vast majority of ionized fragments from the vaporized aerosol in the cold and hot start BA + NO3 experiments (Figures S1p–r and S2p–r) were in the lower m/z range (≤73). The lower m/z peaks at 15 (CH3+), 30 (CH4N+), 44 (C2H6N+), 58 (C3H8N+), and 73 (C4H11N+) are fragments of the unoxidized parent amine. This is consistent with the fragmentation of butylammonium nitrate salt (BA·HNO3). The significant nitrate peaks at m/z 30 (NO+) and 46 (NO2+) also support BA·HNO3 formation. As in the DEA + NO3 experiments, there are higher m/z peaks (>73) at 147 (C4H7N2O4+) and 170 (C6H6N2O4+) that suggest addition chemistry is occurring, possibly forming dimers. There was little change in chemical composition with temperature, consistent with the observations for BA + NO3 aerosol density and volatility. Though the particles completely partition to the gas phase after heating the chamber in the cold start NO3 experiment, the mass spectra suggests that a very small amount of particles return after cooling the chamber.
3.4.4. Organic mass spectra correlation
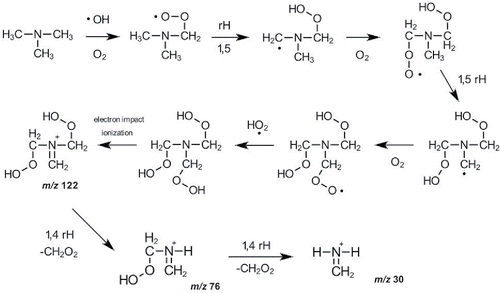
The organic mass spectrum at 10°C was plotted against the organic mass spectrum after heating to 40°C for each of the cold start amine oxidation experiments to highlight the differences in the particle composition (). In the TMA + OH experiment (), the higher m/z fragments from the oligomer compounds are observed to have a slight temperature dependence. The increase of oligomers after heating was due to the release of oligomer precursors from the particle phase, enabling further oligomer formation at higher temperatures. The m/z 76 (C2H6NO2+) and 122 (C3H8NO4+) peaks are significantly reduced by increasing temperature and return after cooling (). A possible mechanism explaining the behavior of these two m/z peaks is shown (). The OH attack commences with a hydrogen abstraction from TMA to produce an alkyl radical followed by addition of molecular oxygen to form a peroxy radical. A 1,5 hydrogen rearrangement subsequently produces a hydroperoxide alkyl radical, which adds a molecular oxygen to form a hydroperoxideperoxy radical followed by another 1,5 hydrogen rearrangement to produce a di-hydroperoxide alkyl radical. Addition of molecular oxygen forms the di-hydroperoxideperoxy radical that reacts with hydroperoxy radical to form a tri-hydroperoxide amine molecule. At cooler temperatures, the tri-hydroperoxide amine molecule partitions to the aerosol phase. Electron impact ionization in the HR-ToF-AMS provides the fragment ion observed at m/z 122. A 1,4 hydrogen rearrangement with the loss of CH2O2 yields m/z 76. An additional 1,4 hydrogen rearrangement with the loss of CH2O2 yields m/z 30. The formation of hydroperoxide amine aerosol at cold temperatures confirms that peroxy and hydroperoxy (RO2· + HO2·) radical chemistry occurs significantly in parallel with the RO2· + RO2· chemistry that leads to oligomer aerosol.
Figure 2. Correlation plots of the organic mass spectra (cold [initial] vs. hot, normalized to total mass) from the cold start (a) TMA + OH, (b) TMA + NO3, (c) DEA + OH, (d) DEA + NO3, (e) BA + OH, and (f) BA + NO3 oxidation experiments. The dashed lines indicate the 1:1 ratio.
![Figure 2. Correlation plots of the organic mass spectra (cold [initial] vs. hot, normalized to total mass) from the cold start (a) TMA + OH, (b) TMA + NO3, (c) DEA + OH, (d) DEA + NO3, (e) BA + OH, and (f) BA + NO3 oxidation experiments. The dashed lines indicate the 1:1 ratio.](/cms/asset/6f8d5019-c7a6-48d3-96e1-04e82172c31e/uast_a_1236182_f0002_oc.gif)
Figure 3. Time profiles for m/z 122 (unit mass resolution) and fragment ion C3H8NO4+ (high resolution) in the cold start (top) and hot start (bottom) trimethylamine with OH oxidation experiments.
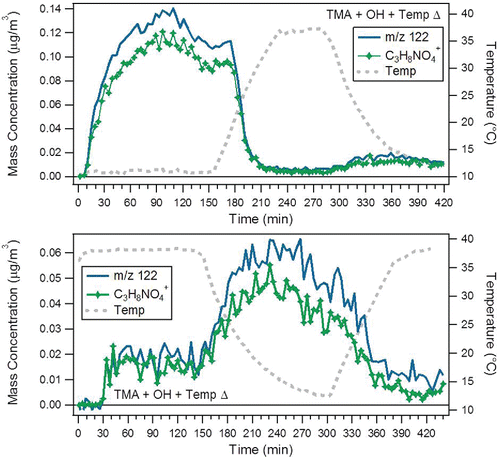
Figure 4. Proposed mechanism for the formation of temperature dependent aerosol with ion fragments at m/z 76 and m/z 122 observed by the HR-ToF-AMS (rH = hydrogen rearrangement).
There are greater amine fragments (m/z 30 and 58) at cold temperature in the DEA + OH experiment (), while there are greater hydrocarbon fragments (m/z 55 and 57) as well as higher m/z peaks at hot temperature. This suggests higher temperatures allow for more gas phase oxidation/combination reactions, resulting in longer carbon chain molecules that partition to the particle phase. The BA + OH experiment correlation () was similar to the DEA + OH correlation in that there was greater hydrocarbon and higher m/z fragments at hot temperature.
The correlation is scattered in the TMA + NO3 experiment () due to gas phase partitioning at higher temperature. In the DEA + NO3 experiment (), the m/z peaks follow the 1:1 line, which means there was little to no change in composition with the increase in temperature. The fraction of total mass in the BA + NO3 experiment () was dominated by lower (<73) m/z fragments. The m/z peaks followed the 1:1 line, indicating no change in composition with the increase in temperature.
3.5. Aerosol chemical composition: Elemental analysis
The HR-ToF-AMS elemental analyses for each of the amine oxidation experiments are shown in . Highly oxidized aerosol was formed in the TMA + OH experiments (oxygen to carbon, O/C > 0.5) with greater oxidation at higher temperatures. The hydrogen to carbon (H/C) ratio decreases at higher temperatures (2.0 → 1.8) as the O/C ratio increases (0.5 → 0.7), suggesting an increase in the number of carbonyl functional groups. The organic mass to organic carbon (OM/OC) ratio also increases at higher temperatures. The OM/OC ratio was very high in the TMA + OH experiments (OM/OC up to 2.5), consistent with the formation of large, highly oxidized oligomer amine compounds. As in the TMA + OH experiments, oxidized aerosol was formed in the DEA + OH experiments (O/C > 0.30) and the BA + OH experiments (O/C > 0.20).
Reduced amine aerosols are formed in the TMA + NO3 experiments (O/C = 0.06, H/C = 2.75, N/C = 0.36), consistent with the formation of TMA·HNO3, as elemental ratios are determined from only the organic portion of the aerosol. Reduced aerosol was also formed in the DEA + NO3 experiments (O/C = 0.04, H/C = 2.88, N/C = 0.48), consistent with the formation of DEA·HNO3. Little to no change in elemental ratio is observed with changes in temperature in the DEA + NO3 oxidation experiments. Reduced aerosol was formed in the BA + NO3 experiments (O/C = 0.02, H/C = 3.00, N/C = 0.55), consistent with the formation of BA·HNO3. Most of the particles were lost with increasing temperature in the cold start BA + NO3 experiment. No change in elemental ratio with temperature change was observed in the hot start BA + NO3 experiment.
4. Conclusion
Temperature had a significant effect on aerosol formed from aliphatic amine precursors. The nucleation temperature is an important factor influencing the overall aerosol formation. In general, more aerosol was formed at colder temperatures. This has important implications for locations influenced by amine emissions during the winter months. Changes in temperature after aerosol formation had an impact on the chemical composition of the aged aerosol through further oxidation of gas phase products released from the aerosol in going from cold to hot, or condensation of gas phase species onto existing aerosol in going from hot to cold. This impact is atmospherically relevant as ambient aerosol experiences temperature gradients due to diurnal cycles or vertical mixing in the atmosphere. The aerosol formed in the TMA + OH oxidation experiments supports the oligomer mechanism discussed in previous work (Price et al. Citation2014). In addition, a novel, temperature dependent, mechanism describing peroxy (RO2·) and hydroperoxy (HO2·) radical reactions was observed in the TMA + OH experiments. Amine peroxy radicals produced from the initial OH attack react with HO2· to form a hydroperoxide compound that partitions to the particle phase at 10°C. The formation of hydroperoxide amine aerosol at cold temperatures confirms that RO2· + HO2· chemistry occurs significantly in parallel with the RO2· + RO2· chemistry that leads to oligomer aerosol.
The aerosol formed in the NO3 oxidation experiments consists mainly of amine nitrate salts. The extent of amine salt formation is highly dependent on temperature and the gas-to-particle equilibrium of the precursor amine. In the temperature range of this study (10°C–40°C), TMA·HNO3 completely partitioned to the gas phase at higher temperatures while DEA·HNO3 did not. The empirical observations of the gas-to-particle partitioning of TMA·HNO3 and DEA·HNO3 are consistent with solid/gas dissociation constants determined by Ge et al. (Citation2011b). The high concentrations of amine salt aerosol observed in these experiments (100–600 μg/m3) underlines the importance of amine reactions in the atmosphere. This is consistent with ambient measurements of amine aerosol in wintertime (Tan et al. Citation2002; Silva et al. Citation2008) when ambient temperatures can be 20–30°C colder than the minimum temperature investigated in this study. Amine nitrate salts are also an important contributor to aerosol formation during the nighttime hours, when temperatures tend to be cooler.
UAST_1236182_Supplemental_File.zip
Download Zip (357 KB)Related Research Data
References
- Barsanti, K. C., McMurry, P. H., and Smith, J. N. (2009). The Potential Contribution of Organic Salts to New Particle Growth. Atmos Chem. Phys., 9:2949–2957.
- Borduas, N., Abbatt, J. P. D., and Murphy, J. G. (2013). Gas Phase Oxidation of Monoethanolamine (MEA) with OH Radical and Ozone: Kinetics, Products, and Particles. Environ Sci Technol., 47:6377–6383.
- Cadle, S. H., and Mulawa, P. A. (1980). Low-Molecular Weight Aliphatic-Amines in Exhaust from Catalyst-Equipped Cars. Environ. Sci. Technol., 14:718–723.
- Carter, W. P. L., Cocker, D. R., III, Fitz, D. R., Malkina, I. L., Bumiller, K., Sauer, C. G., Pisano, J. T., Bufalino, C., and Song, C. (2005). A new Environmental Chamber for Evaluation of Gas-Phase Chemical Mechanisms and Secondary Aerosol Formation. Atmos Environ., 39:7768–7788.
- Clark, C. H., Kacarab, M., Nakao, S., Asa-Awuku, A., Sato, K., and Cocker, D. R. (2016). Temperature Effects on Secondary Organic Aerosol (SOA) from the Dark Ozonolysis and Photo-Oxidation of Isoprene. Environ. Sci. Technol., 50:5564–5571.
- Cocker, D. R., Flagan, R. C., and Seinfeld, J. H. (2001). State-of-the-Art Chamber Facility for Studying Atmospheric Aerosol Chemistry. Environ. Sci. Technol., 35:2594–2601.
- DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L. (2006). Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer. Anal Chem., 78:8281–8289.
- Dockery, D. W. (2009). Health Effects of Particulate Air Pollution. Ann Epidemiol., 19:257–263.
- Ehara, K., Hagwood, C., and Coakley, K. J. (1996). Novel Method to Classify Aerosol Particles According to their Mass-to-Charge Ratio—Aerosol particle Mass Analyser. J. Aerosol Sci., 27:217–234.
- Ge, X., Wexler, A. S., and Clegg, S. L. (2011a). Atmospheric Amines – Part I, A Review. Atmos Environ., 45:524–546.
- Ge, X. L., Wexler, A. S., and Clegg, S. L. (2011b). Atmospheric Amines - Part II. Thermodynamic Properties and Gas/Particle Partitioning. Atmos Environ., 45:561–577.
- Griffin, R. J., Cocker, I., , D.R., Flagan, R. C., and Seinfeld, J. H. (1999). Organic Aerosol Formation from the Oxidation of Biogenic Hydro- Carbons. J. Geophys. Res., 104:3555–3567.
- IPCC (2013). Climate change 2013: The physical science basis. Contribution of working group I to the fifth assessment report of the Intergovernmental Panel on Climate Change, T.F. D. Qin, G.-K. Plattner, M. Tignor, S.K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex, and P.M. Midgley, eds., Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, pp. 1535.
- Jayne, J. T., Leard, D. C., Zhang, X. F., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R. (2000). Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles. Aerosol Sci. Technol., 33:49–70.
- Lee, D., and Wexler, A. S. (2013). Atmospheric Amines - Part III: Photochemistry and Toxicity. Atmos Environ., 71:95–103.
- Malloy, Q. G. J., Li, Q., Warren, B., Cocker, D. R., III, Erupe, M. E., and Silva, P. J. (2009a). Secondary Organic Aerosol Formation from Primary Aliphatic Amines with NO3Radical. Atmos Chem. Phys., 9:2051–2060.
- Malloy, Q. G. J., Nakao, S., Qi, L., Austin, R., Stothers, C., Hagino, H., and Cocker, D. R. (2009b). Real-Time Aerosol Density Determination Utilizing a Modified Scanning Mobility Particle Sizer Aerosol Particle Mass Analyzer System. Aerosol Sci. Technol., 43:673–678.
- Mosier, A. R., Andre, C. E., Viets, F. G., Jr. (1973). Identification of Aliphatic Amines Volatilized from Cattle Feedyard. Environ. Sci. Technol., 7:642–644.
- Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H. (1996). Gas/Particle Partitioning and Secondary Organic Aerosol Yields. Environ. Sci. Technol., 30:2580–2585.
- Pope, C. A., Thun, M. J., Namboodiri, M. M., Dockery, D. W., Evans, J. S., Speizer, F. E., and Heath, C. W. (1995). Particulate Air-Pollution as a Predictor of Mortality in a Prospective-Study of Us Adults. Am. J. Respir. Crit. Care Med., 151:669–674.
- Price, D. J., Clark, C. H., Tang, X. C., Cocker, D. R., Purvis-Roberts, K. L., and Silva, P. J. (2014). Proposed Chemical Mechanisms Leading to Secondary Organic Aerosol in the Reactions of Aliphatic Amines with Hydroxyl and Nitrate Radicals. Atmos Environ., 96:135–144.
- Qi, L., Nakao, S., Tang, P., and Cocker, D. R. (2010). Temperature Effect on Physical and Chemical Properties of Secondary Organic Aerosol from M-Xylene Photooxidation. Atmos Chem. Phys., 10:3847–3854.
- Silva, P. J., Erupe, M. E., Price, D. J., Elias, J., Malloy, Q. G. J., Li, Q., Warren, B., and Cocker, D. R., III (2008). Trimethylamine as Precursor to Secondary Organic Aerosol Formation via Nitrate Radical Reaction in the Atmosphere. Environ. Sci. Technol., 42:4689–4696.
- Smith, J. N., Dunn, M. J., VanReken, T. M., Iida, K., Stolzenburg, M. R., McMurry, P. H., and Huey, L. G. (2008). Chemical Composition of Atmospheric Nanoparticles Formed from Nucleation in Tecamac, Mexico: Evidence for an Important Role for Organic Species in Nanoparticle Growth. Geophys. Res. Lett., 35.
- Takekawa, H., Minoura, H., and Yamazaki, S. (2003). Temperature Dependence of Secondary Organic Aerosol Formation by Photo-Oxidation of Hydrocarbons. Atmos Environ., 37:3413–3424.
- Tan, P. V., Evans, G. J., Tsai, J., Owega, S., Fila, M. S., and Malpica, O. (2002). On-Line Analysis of Urban Particulate Matter Focusing on Elevated Wintertime Aerosol Concentrations. Environ. Sci. Technol., 35:3512–3518.
- Tang, X., Price, D., Praske, E., Vu, D. N., Purvis-Roberts, K., Silva, P. J., Cocker Iii, D.R., and Asa-Awuku, A. (2014). Cloud Condensation Nuclei (CCN) Activity of Aliphatic Amine Secondary Aerosol. Atmos Chem. Phys., 14:5959–5967.
- Warren, B., Austin, R. L., and Cocker, D. R. (2009). Temperature Dependence of Secondary Organic Aerosol. Atmos Environ., 43:3548–3555.
- Watson, J. G. (2002). Visibility: Science and Regulation. J Air Waste Manage, 52:628–713.
- Westerholm, R., Li, H., Almen, J. (1993). Estimation of Aliphatic Amine Emissions in Automobile Exhausts. Chemosphere, 27:1381–1384.