
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Light-absorbing organic aerosol (brown carbon, BrC) impacts the radiative balance of the earth’s atmosphere; however, the magnitude of this impact is poorly constrained due to uncertainties in BrC sources, composition, and lifetime. In particular, the role of chemical “aging” on the optical properties of BrC particles is poorly understood. Here we carry out laboratory studies aimed at understanding how one such aging process, heterogeneous oxidation, may affect the chemical and optical properties of biomass burning-derived BrC. We generate BrC from smoldering ponderosa pine needles, oxidize the BrC in a flow reactor, and use simultaneous measurements of aerosol optical properties and chemical composition to monitor changes upon oxidation. Under the set of conditions investigated here, we find that with increased oxidant exposure, the aerosol becomes more oxidized and less absorbing, presumably due to oxidative degradation of the chromophores. Both the kinetics and evolution of this process are oxidant dependent. While heterogeneous oxidation by ozone results in a rapid “bleaching” of the BrC (i.e., decrease in absorptivity), a substantial fraction of the BrC is resistant to bleaching by this mechanism. In contrast, bleaching due to heterogeneous oxidation by OH in the presence of ozone remains active over long timescales (timescale of days), suggesting a sustained evolution of BrC optical properties throughout the aerosol atmospheric lifetime.
Copyright © 2019 American Association for Aerosol Research
1. Introduction
Atmospheric aerosol particles can directly impact the Earth’s radiative balance via the scattering and absorption of radiation (“aerosol–radiation interactions”) (Stocker et al. Citation2013). As organic aerosol (OA) is a major component of submicron aerosol mass (Zhang et al. Citation2007), understanding its optical properties is critical for calculating aerosol radiative forcing (Stocker et al. Citation2013). Due to the often weak absorption properties of many organic species (compared to strong absorbers such as black carbon and mineral dust), OA is traditionally considered to predominantly scatter light (Seinfeld and Pandis Citation2006). However, some components of OA may absorb UV and visible light; this fraction, known as brown carbon (BrC), may result in warming, somewhat offsetting the cooling (scattering) effect of OA (Stocker et al. Citation2013). Global model simulations have shown that BrC can indeed be an important contributor to overall aerosol radiative forcing (Chung, Ramanathan, and Decremer Citation2012; Feng, Ramanathan, and Kotamarthi Citation2013; Jo et al. Citation2016; Lin et al. Citation2014; Saleh et al. Citation2015; Wang et al. Citation2014; Wang et al. Citation2018). However, such contributions, in both the present and future atmospheres, remain highly uncertain, due to the high level of chemical complexity and variability of BrC aerosols. Organic compounds contributing to light absorption likely comprise a small mass fraction of total OA, and their chemical identity is complex and uncertain (Laskin, Laskin, and Nizkorodov Citation2015). Furthermore, despite considerable study in recent years, virtually all aspects of the BrC lifecycle and impacts (sources, composition, properties, and lifetime) remain poorly constrained (Laskin, Laskin, and Nizkorodov Citation2015).
Biomass and biofuel burning, an important global source of OA in general (Bond et al. Citation2004; Hallquist et al. Citation2009), is also an important atmospheric source of primary BrC (Laskin, Laskin, and Nizkorodov Citation2015). With future increases in biomass burning emissions projected (Pechony and Shindell Citation2010), constraining biomass burning BrC is important for projections of radiative forcing. Understanding the climate impacts of primary biomass burning BrC, however, requires not only knowledge of the emissions and properties and BrC particles, but also knowledge of their atmospheric lifetimes. Examples of biomass burning BrC chromophores include polycyclic aromatic hydrocarbons, phenols, aromatic acids, and nitroaromatic compounds (Lin et al. Citation2017; Lin et al. Citation2018; Bluvshtein et al. Citation2017; Mohr et al. Citation2013). As atmospheric processing is known to alter the chemical composition of OA (Jimenez et al. Citation2009), it is likely that BrC chromophores will be altered as well. Various aging processes could potentially make the BrC more absorbing (“browner”) or less absorbing (“whiter”), and thus it may not be appropriate to assign a single set of optical properties to BrC over its entire atmospheric lifetime. Furthermore, if whitening/bleaching occurs, the lifetime of biomass burning-derived primary BrC may be substantially different from the lifetime of biomass burning OA overall.
Indeed, field measurements of ambient BrC in biomass burning plumes support the view of BrC as a dynamic system. Studies tracking individual plumes find that light absorption by BrC drops rapidly after emission, with lifetimes on the order of a single day (Adler et al. Citation2011; Forrister et al. Citation2015; Wang et al. Citation2018), suggesting substantial bleaching with atmospheric age. However, these studies also indicate a persistent lower level of absorption over longer timescales, indicating that such bleaching processes do not necessarily proceed to completion (Forrister et al. Citation2015; Wang et al. Citation2018).
In recent years, a number of laboratory studies have examined the effects of various aging processes on absorption by biomass burning-derived BrC. These include photooxidation (Chang and Thompson Citation2010; Gelencsér et al. Citation2003; Hems and Abbatt Citation2018; Zhao et al. Citation2015) and photolysis (Wong, Nenes, and Weber Citation2017; Zhao et al. Citation2015) of bulk aqueous solutions containing BrC species; and photooxidation of complex multiphase (particle and gas) biomass burning mixtures (Saleh et al. Citation2013; Zhong and Jang Citation2014). All experiments found changes to light absorption with oxidation, though the nature of the change (browning, bleaching, or one followed by the other) varies with aging process and chemical system studied.
These previous studies have focused primarily on how chemical transformations within the particle can affect BrC absorption. There has been considerably less direct study on how heterogeneous oxidation reactions may affect light absorption properties of BrC particles. The only exceptions are recent studies by Sumlin et al. (Citation2017) and Li et al. (Citation2019). Sumlin et al. (Citation2017) exposed particles generated from smoldering peat to high levels of ozone and OH and found small decreases to light absorption from heterogeneous oxidation by OH (the effects of ozone was not quantified), though such changes were found to occur only after the equivalent of several days of atmospheric oxidation. Likewise, Li et al. (Citation2019) oxidized tar balls by exposure to high levels of OH and ozone and found bleaching to occur. These results stand in contrast to the more immediate changes observed in field studies (Forrister et al. Citation2015; Xuan Wang et al. Citation2018).
In this work, we investigate further the effects of heterogeneous oxidation on the absorption of biomass burning-derived BrC by adding additional constraints on oxidation by OH and also investigating the role of ozone. BrC is generated from the smoldering of ponderosa pine needles, which generates OA and BrC but no black carbon (soot). The generated particles are then sent into an oxidation flow reactor to simulate hours to days of atmospheric oxidation, first by ozone alone and then by ozone and OH. A particular focus of this work is the kinetics of the changes to absorption properties of the BrC. This focus provides context for how heterogeneous oxidation, under the conditions studied here, may affect BrC optical properties during atmospheric transport.
2. Methods
Experiments, carried out at the Advanced Light Source (ALS) at Lawrence Berkeley National Laboratory (Berkeley, CA, USA), involved a BrC generator to form biomass burning particles, an oxidation flow reactor to heterogeneously oxidize the particles, and several analytical instruments to measure the particles’ evolving chemical and optical properties. The BrC generator consisted of a hot plate inside a steel container (30.48 cm × 25.4 cm × 25.4 cm) with a glass lid. A strip of silicone between the lid and the steel container, and a weight on the glass lid, served to seal the box and prevent leaks. The temperature of the hot plate surface, monitored with a thermocouple, was held at ∼300 °C. During experiments, BrC was generated in real time and flushed from the chamber with a stream of N2. By controlling the temperature and eliminating O2 from the system, biomass fuel can be smoldered with no flaming combustion, thus generating BrC aerosol in the absence of soot/black carbon (BC) formation (Chen and Bond Citation2010). The absence of BC simplifies the interpretation of optical property evolution as no assumptions regarding mixing state and BC optical properties are required.
The fuel used in these experiments was pine needle litter, collected from the base of a ponderosa pine tree near the ALS. Ponderosa pine was chosen as it is a common biomass burning fuel in the Western United States. Only the top layer of litter was collected as this layer contained less moisture than the layers near the ground. In a given experiment, a bundle of needles (∼0.5 cm in diameter) was placed in an aluminum foil boat on the hot plate and the lid to the generator was sealed. To reach an approximate target initial aerosol output, the quantity of pine needles was typically adjusted during the first few minutes of a smoldering event. Aerosols and gases produced by the smoldering event were flushed from the box with 3.1 slpm flow of N2. This flow rate was necessary to limit the size of the particles to ∼100–200 nm mean surface area-weighted diameter. The volume-weighted concentration of the aerosol was the highest at the beginning of each smoldering event, and decayed exponentially over the course of the smoldering. Although some experimental difficulties resulted from the temporal inhomogeneity of the aerosol generation, such as the occasional clogging of lines and the variable aerosol loading (described below), this flow-through method allowed for authentic combustion-derived aerosol to be studied without modification, thereby avoiding potential artifacts that might arise from collection or handling of samples. For instance, the aerosol retained water-soluble and -insoluble compounds, which might otherwise be removed if the aerosol were collected on a filter, extracted then resuspended in solvent, and reaerosolized.
Only a small fraction (100 sccm) of the flow from the BrC generator was sent into the flow reactor for heterogeneous oxidation, with the remainder (∼3 slpm) sent to exhaust. The sample flow was passed through a charcoal denuder to remove gaseous species. This flow was subsequently combined with an 820 sccm carrier flow of ∼24% O2 in N2 at ∼37% relative humidity (RH), a 50 sccm flow of variable ozone concentration in an N2/O2 mixture, and a 25 sccm mixture of ∼5 ppm hexane in N2. This mixture (total flow of ∼1 slpm, with an O2 mixing ratio of ∼20% and 30% RH) was then sent into the oxidation flow reactor, described in detail by Smith et al. (Citation2009). Briefly, the reactor was a quartz flow tube (130 cm long, 2.5 cm inner diameter) surrounded by four 254 nm lights; these remained off for the ozone-only experiments and are turned on to generate OH via the photolysis of ozone in the presence of water vapor. Total residence time in the reactor was ∼38 s. OH concentrations were monitored by sampling a small flow through a potassium iodide filter to remove ozone and into a gas chromatograph with a flame ionization detector (GC-FID). The GC-FID was used to measure the decay of the added hexane and thus determine the OH exposure using mixed-phase relative rate kinetics (Smith et al. Citation2009). Experimental conditions, including the range of oxidant exposures investigated and aerosol concentration and diameter, are provided in Table S1 for the experiments discussed here.
Flow from the reactor was sent through a Carulite (Carus Corporation) denuder to remove ozone prior to being sampled by the aerosol instruments. Chemical composition of the aerosol was measured in real time using an Aerodyne high-resolution time-of-flight aerosol mass spectrometer (DeCarlo et al. Citation2006), using vacuum ultraviolet (VUV) single-photon ionization at 10.5 eV from the Chemical Dynamics Beamline (9.0.2) at Lawrence Berkeley National Laboratory (Northway et al. Citation2007). The low photon energy used in VUV generally results in less fragmentation than electron ionization (EI), thus providing more detailed molecular information than in the standard (EI) AMS operation mode. However, with VUV ionization, oxygen-containing organic ions have a higher propensity for fragmentation than reduced organic ions (Kroll et al. Citation2012; Smith et al. Citation2009). This fragmentation complicates molecular characterization of the BrC aerosol and its oxidation products. The exact chemical composition of ions with m/z ≤ 126 was identified by high-resolution analysis. Analysis of higher m/z ions was typically restricted to unit mass resolution.
The remaining flow was diluted with make-up particle-free N2, necessary because of sampling requirements of the other instruments, and passed through a Carulite denuder to remove ozone. A scanning mobility particle sizer (TSI model 3080) was used to quantify particle number and electrical mobility diameter, and the UC Davis cavity ring-down spectrometer and photo-acoustic spectrometer (CRD/PAS) (Cappa et al. Citation2011; Lack et al. Citation2012; Langridge et al. Citation2011) was used to measure aerosol optical properties. Briefly, the CRD/PAS measures extinction and absorption of dry aerosol at 405 nm and 532 nm as well as the 532 nm extinction of aerosols conditioned to both 75% and 85% RH. For this work, we consider only absorption by the dry aerosol. The PAS was calibrated relative to the CRD using gas-phase ozone and NO2 (Cappa et al. Citation2011). A filtered channel on the instrument is used to continuously monitor for drift in any gas-phase background that may affect the absorption. This channel confirmed no drift in the background during these experiments. Estimated uncertainties are 4% for the extinction measurements and 8% for the absorption measurements, as determined from the calibration uncertainties (Cappa et al. Citation2012; Lack et al. Citation2012; Langridge et al. Citation2011).
Effective complex refractive indices (RI; m = n−ik) were retrieved using an optical closure method (Lambe et al. Citation2013; Zarzana, Cappa, and Tolbert Citation2014). In brief, the difference between the observed absorption (babs) and extinction (bext) coefficients and those calculated from spherical particle Mie theory assuming homogeneous particles using the observed particle size distribution as input is minimized by varying both the real and imaginary parts of the RI. A fuller description is provided in the online supplementary information (SI). The imaginary component (k) is broadly reflective of the material absorptivity. For the measurements here, we have estimated the uncertainty in the k values via a Monte Carlo analysis as ±0.0001 at 532 nm and ±0.0005 at 405 nm, corresponding to ∼±15%, consistent with Zarzana, Cappa, and Tolbert (Citation2014) (see SI). However, the precision is determined by instrument stability and is likely better for a given experiment, at least at 405 nm. For a 5-min average, we estimate the precision to be on the order of 0.0001. The absorption Ångström exponent (AAE), which characterizes the wavelength dependence of absorption, was calculated from the measured absorption at 405 and 532 nm by
(1)
(1)
Incomplete removal of gases by the denuder or dilution-induced aerosol evaporation may lead to the presence of gas-phase species that, upon oxidation, may repartition to the aerosol influencing composition and optical properties via a gas-phase (nonheterogeneous) process. We investigate this possibility by analyzing how step changes in oxidation conditions alter the total aerosol number concentration. Oxidation of gas-phase compounds would presumably increase the total aerosol number through nucleation of new particles. Specifically, we investigate how aerosol number is altered by turning the lights (which generate OH) on or off in the presence of constant ozone concentrations. In experiments without the denuder present (in the presence of gas-phase compounds), we observed step-function changes in the particle number with the particle number decreasing by an order of magnitude when the lights were turned off and increasing back to the previous level when the lights were turned back on. When the denuder was present, no such changes were observed. Although new particle formation is an imperfect proxy for nonheterogeneous processes, the difference between the denuded and nondenuded experiments suggests that changes resulting from heterogeneous oxidation dominate in the experiments using the denuder.
3. Results and discussion
3.1. Characteristics of fresh (unoxidized) aerosol
The unoxidized aerosol formed from the smoldering of ponderosa pine needles is chemically complex, as indicated by the observed mass spectra (Figure S1). As expected when using VUV ionization, a soft ionization technique, the mass spectra exhibit high-intensity ions over a wide range of masses including significant peaks at m/z > 300. We have tentatively identified two of these high mass peaks—abietic acid (m/z 302; C20H30O2+) and β-sitosterol (m/z 414; C29H50O+)—both of which have been previously measured in aerosol from emitted from the burning of ponderosa pine (Oros and Simoneit Citation2001). At lower m/z, where we perform high-resolution identification of the ions, the mass spectrum likely includes fragment ions. As described above, oxygenates exhibit increased fragmentation compared to reduced species; this effect at least partly accounts for the large abundance (>50% of the signal analyzed in high resolution) of oxygenated ions observed at m/z < 126, such as C2H4O2+ and C3H5O2+. In AMS measurements using electron ionization, enhanced intensities of C2H4O2+ and C3H5O2+ are frequently observed in aerosol influenced by biomass burning. The ions are associated with anhydrosugars such as levoglucosan, formed by the thermal degradation of cellulose and hemicellulose (Alfarra et al. Citation2007; Schneider et al. Citation2006). Organic nitrogen compounds are notably absent from the signal, with their abundance always <0.5% of the total intensity identified by high-resolution analysis. The low nitrogen content is consistent with other measurements of the bulk composition of biomass burning OA generated from ponderosa pine fuel (Ortega et al. Citation2013). For the analysis presented here, we focus on ensemble properties of the composition, defining ion classes using the high-resolution assignments of the low mass ions. To reduce the effect that oxygenate fragmentation may have on the interpretation of the evolution of reduced species, we consider small (C1–C5) and large (C6–C9) CxHy+ ions separately.
The aerosols generated in this process are mildly absorbing at 405 nm, with values of k405 (the imaginary component of the effective refractive index at 405 nm) ranging from 0.001 to 0.006. These values correspond to a range of mass absorption coefficients of ca. 0.02–0.14 m2 g−1 (assuming 200-nm-diameter particles with a density of 1.3 g cm−3). The measured k405 values fall at the low end of reported values of measured values for both ambient biomass burning particles (Lack et al. Citation2012; Zhang et al. Citation2016) and laboratory-generated smoldering aerosols (Chakrabarty et al. Citation2010; Sumlin et al. Citation2017), which generally range from ∼0.005 to ∼0.04. Such differences are likely attributable to differences in combustion conditions, assumptions regarding aerosol mixing state and black carbon content, and the specific fuels. For instance, flaming combustion would have caused higher black carbon-to-OA ratios, which have been shown to correspond to more strongly absorbing OA (Saleh et al. Citation2014). However, despite the relatively low level of light absorption by the freshly emitted particles in the present experiment, we are still able to determine the kinetics of changes to the aerosol absorption upon heterogeneous oxidation.
This work uses authentic biomass samples, generated in and sampled in situ, which inherently introduces variability into the experiment. Although the pine needles were collected at an identical time and location and smoldered under the same conditions, we observed variability in the composition and optical properties of the fresh (unoxidized) aerosol. The composition of emitted particles varied from one smoldering event to another (Figure S1); to minimize the effect of burn-to-burn variability on results, we focus on two smoldering events for which we were able to take measurements at multiple oxidation exposures using one sample. As the fuel may undergo chemical and physical changes as it is heated over the course of the ∼20–60 min experiment, we investigated this source of variability in a series of control experiments. The observed changes (Figures S2 and S3) are minor and occur primarily at the beginning of a smoldering event. Consideration of data acquired at least 10 min after the start of a smoldering event ensures that the magnitude of nonoxidative changes is small relative to those observed upon oxidation (described below). Additionally, in the ozone + OH experiment, the OH exposure was altered nonmonotonically to further reduce the influence of drift. As shown in Figure S4, the trends in composition and absorption follow OH exposure. Overall, these results suggest that the changes to particle composition and optical properties observed over the course of our aging experiments are a result of oxidation, and not from changes to the fresh particles or other nonoxidative processes.
3.2. Heterogeneous oxidation by ozone
Changes to the composition and optical properties of the particles upon exposure to gas-phase ozone are shown in . Heterogeneous oxidation by ozone leads to formation of more oxidized molecules within the particles, as evidenced by the decrease in the relative abundance of CxHy+ ions and the increase in the relative abundance of CxHyOz+ ions with ozone exposure (). These chemical changes are accompanied by corresponding decreases in the retrieved k of the particles (at both 405 nm and 532 nm), indicating lowered absorptivity as shown in . The changes observed are ultimately small (e.g., k405 decreases from 0.004 to 0.003), but non-negligible. Such changes likely result from ozonolysis reactions of conjugated alkene molecules within the particles; by reducing the extent of conjugation, the chromophores will degrade, shifting the light absorption to shorter wavelengths and decreasing the absorption of the particle at the longer wavelengths. No systematic change as a function of oxidation is observed for the retrieved n of the particles. The averaged values are n405 = 1.53 ± 0.01 and n532 = 1.49 ± 0.006, where the uncertainties are the standard deviation of the retrieved n across all oxidation levels.
Figure 1. Changes in (a) aerosol composition, (b) effective imaginary refractive index, and (c) AAE as a function of ozone exposure. At increased exposures, the aerosol is observed to become more oxidized and less absorbing. Atmospheric age is calculated assuming an ozone mixing ratio of 40 ppbv and is shown on the top axis. The error bars represent the standard deviation of the measurement during each oxidation step. Only one retrieval of k was made for each oxidation step and thus no error bars are reported for k405 and k532. Precision-based error bars, using an estimated precision of ∼0.0001, would be smaller than the symbols.
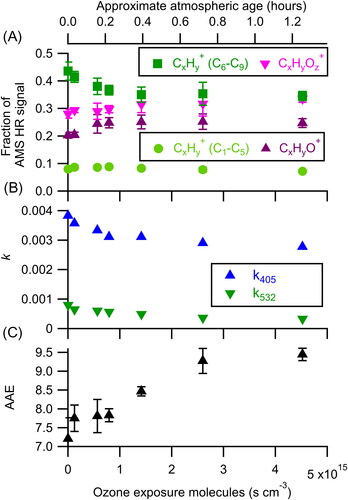
Both the compositional and optical properties of the aerosol plateau at ozone exposures of ∼2.6 × 1015 molecules s cm−3. Assuming an ambient ozone concentration of 40 ppbv, this exposure corresponds to an atmospheric processing time of just under 45 min, suggesting that this observed initial bleaching would occur extremely rapidly in the atmosphere. The nonzero plateau value for k405, however, indicates that a fraction of the BrC is more persistent. This persistence may result from a number of effects, including the depletion of molecules/chromophores that react with ozone, mass transfer limitations due to changing aerosol characteristics, and/or the possibility that the ozone uptake follows Langmuir–Hinshelwood kinetics (George and Abbatt Citation2010). With the current data, we are unable to unambiguously determine the reason for this plateauing. Regardless of the exact underlying mechanism, these results suggest that ozone is unable to completely “bleach” BrC particles.
A second noticeable feature of ozone oxidation is the increase in AAE as oxidation progresses (). This increase is the result of the faster decay of absorption at 532 nm compared to 405 nm () which may result from either a difference in the reactivity toward ozone of the molecules that contribute to absorption at the two wavelengths, or the possibility that molecules may be only partially bleached after one reaction. For instance, oxidation may reduce the extent of conjugation such that the molecule no longer absorbs at longer wavelengths while still absorbing at shorter wavelengths. Alternatively, or in addition, it could indicate a difference in the AAE between the light-absorbing molecules that are reactive toward O3 and those that are not.
Figure 2. Fraction of the original signal remaining as a function of ozone exposure. The fastest decaying CxHy+ ions refer to the three most rapidly decaying high-resolution ions (C5H6+, C7H9+, and C8H9+). While k405 decays at a rate similar to the C6–C9 CxHy+ ions, k532 decays more quickly approaching the rate of the most rapidly decaying ions. Atmospheric age is calculated assuming an ozone mixing ratio of 40 ppbv and is shown on the top axis.
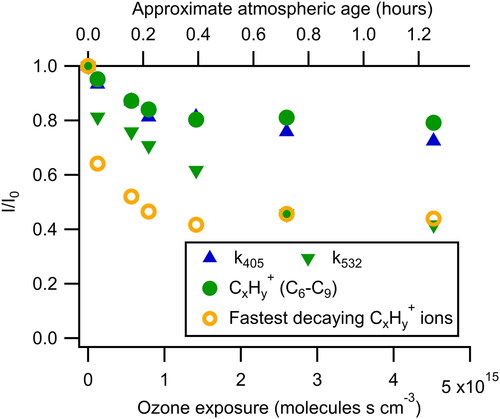
The simultaneous optical and compositional measurements of this study provide insight into how the reactivity of the specific compounds contributing to absorption compare to the overall reactivity of the aerosol. As shown in , k405 decreases at a rate similar to the C6–C9 CxHy+ ions, while k532 decays at a rate more similar to the three most rapidly decaying high-resolution ions (C5H6+, C7H9+, and C8H9+). Some other higher mass ions, measured at unit mass resolution, appear to decay even more quickly than these three, but their low intensities lead to increased uncertainty in their decay kinetics. Overall, these results suggest that the molecules that absorb at longer wavelengths are also those that are most reactive to ozone, and that a substantial fraction of the absorbing molecules within BrC are either nonreactive with ozone or shielded from the reaction.
3.3. Heterogeneous oxidation by OH + ozone
Results from the photooxidation experiment, in which particles are exposed to gas-phase OH radicals, are shown in . As the generation of OH requires the presence of ozone, both oxidants are present during these experiments. Sufficient aerosol output from the smoldering apparatus limited the total experiment time to ∼60 min. The time required for the hexane measurement by GC-FID (∼15 min) limited the number of oxidation steps that could be measured for a given sample. Thus, no measurement of the fresh (unoxidized) particles were made and the composition and optical properties of the particles were instead measured at relatively high OH exposures, ranging from 2.65 × 1011 to 6.75 × 1011 molecules s cm−3, which corresponds to 2–5.2 days of equivalent atmospheric oxidation (assuming an average atmospheric OH concentration of 1.5 × 106 molecules cm−3). The advantage of running at these high oxidant exposures is that it minimizes the influence of oxidation by ozone (which is present in all experiments) on observed results, as such chemistry will have gone to completion by this point (assuming that synergistic effects from the presence of multiple oxidants are negligible). The minimum ozone exposure in these experiments is 3.2 × 1015 molecules s cm−3, which is greater than the value where the plateau in the ozone-only experiment was observed (). The maximum ozone exposure is 1.8 × 1016 molecules s cm−3. Additionally, the impact of 254 nm radiation used for the ozone photolysis is investigated through a lights-only experiment, without ozone (Figure S3), and is determined to have a negligible impact on the interpretation of the oxidation results. This is consistent with the conclusion of Malecha, Cai, and Nizkorodov (Citation2018) that exposure to 254 nm radiation in oxidation flow reactors leads to negligible mass loss. Thus, it is reasonable to assume that the changes observed during these OH exposure variations are the result of OH rather than ozone chemistry or photolysis. We make no corrections to the data for potential changes resulting from either process.
Figure 3. Changes in (a) aerosol composition, (b) effective imaginary refractive index, and (c) AAE as a function of OH + ozone exposure. The solid lines represent an exponential fit used to determine the effective second order rate constants for C6–C9 CxHy+ (7.4 × 10−13±0.7 × 10−13 cm3 molecules−1 s−1) and k405 (9 × 10−13±2 × 10−13 cm3 molecules−1 s−1). The error bars represent the standard deviation of the measurement during each oxidation step. Atmospheric age is calculated assuming an OH concentration of 1.5 × 106 molecules cm−3.
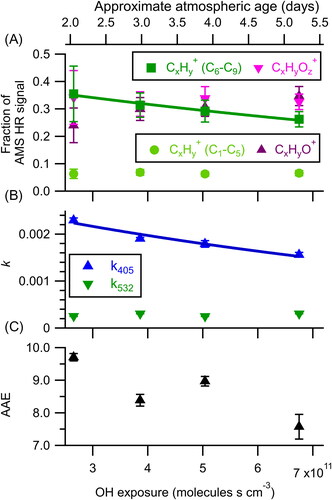
shows how chemical composition of the aerosol was affected by OH exposure. Changes are qualitatively similar to those in the ozone experiment (), with reduced ions (CxHy+) decreasing and oxidized ions (CxHyO+ and CxHyOz+) increasing with OH exposure. Changes to optical properties are shown in . As in the ozone case, k405 decreases with oxidation, indicating the oxidative degradation of the chromophores within the BrC. By contrast, the k532 displayed a negligible trend with changing OH exposures, though k532 was already extremely low (∼2.5 × 10−4) by the first oxidation step. As a result, the AAE value decreases at increasing exposure levels, consistent with the ambient observations reported by Forrister et al. (Citation2015). Similar to the ozone-only experiment, we observe no systematic change in the retrieved n. Average retrieved values are n405=1.57 ± 0.01 and n532=1.51 ± 0.003, where the uncertainties are the standard deviation of the retrieved n across all oxidation levels.
As we did not measure the composition or optical properties of the nonoxidized particles in this experiment, we cannot assess how these quantities relate or change early in their lifetime in the atmosphere (first ∼2 days after emission). However, the kinetics of subsequent changes can be estimated from exponential fits to the absolute values (solid lines in ). In this analysis, we assume that ozonolysis contributes negligibly to the observed changes over the course of the OH experiment and that bleaching follows second-order kinetics. Within the errors of the fits, the effective second-order rate coefficient of k405 ((9 ± 2)×10−13 cm3 molecules−1 s−1) and of the C6–C9 CxHy+ ions ((7.4 ± 0.7)×10−13 cm3 molecules−1 s−1) are the same. While the C6–C9 CxHy+ decay is likely influenced by molecules that are both absorbing and nonabsorbing, the agreement in the decay rates suggests that chemical composition, in particular reduced, higher molecular weight ions, may provide information on chromophore abundance.
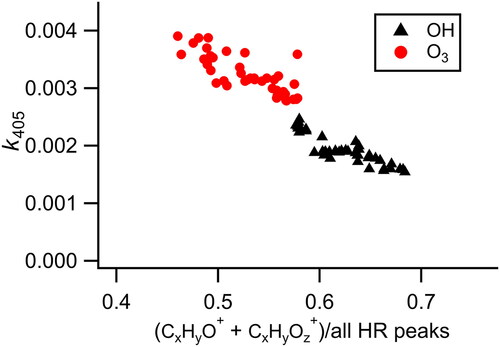
For a system such as this where the chromophores are likely conjugated systems, oxidation will ultimately result in a loss of conjugation as functional groups are added and carbon–carbon double bonds broken. The effect of the initial steps of oxidation is more difficult to predict as the extent of conjugation will depend on the specific functional groups produced in the reaction. Overall, it is expected that there will exist an anticorrelation between absorption and oxygen content, consistent with the results observed here (). However, the complex nature of BrC composition and aging suggest that the exact trend observed will be specific to a given sample/aging chemistry. Indeed, when combining the present results with measurements of other smoldering events (Figure S5), we were unable to find any consistent clear relationships between absorption and key chemical parameters measured, including the normalized intensity of various ions, elemental ratios of the HR peaks, fraction of the signal occurring at high mass (defined using various cutoffs ranging from m/z 126 to >300), and the aromaticity index. The lack of a robust correlation is perhaps unsurprising given the variability of aerosol composition and the expectation that the absorbing species account for a minor fraction of the overall aerosol mass. Overall, we conclude that while absorption generally decreases with increasing oxygen content, ensemble composition measurements are unable to robustly predict absorption.
Figure 4. Relationship between k405 and the aerosol oxygen content during heterogeneous oxidation by ozone and OH + ozone showing a connection between increasing oxygen content of the aerosol and decreased absorption. For completeness, measurements during transitions between oxidant levels (which are not used in the kinetics analysis, ) in the flow tube are included here.
3.4. Bleaching kinetics
The results presented here suggest that heterogeneous oxidation of BrC generated from smoldering events leads to wavelength-dependent bleaching of the particles. Bleaching from reaction with ozone occurs quickly and plateaus on the timescale of an hour. For this sample, bleaching ultimately leads to relatively small changes in the absorptivity. During this process, the AAE is observed to increase with age due to faster decay of chromophores absorbing at longer wavelengths than at shorter wavelengths, suggesting that the assumption that AAE decreases with bleaching may not always hold. The increase in AAE with oxidation by ozone contrasts with field observations that showed a decrease in AAE with increased atmospheric processing (Forrister et al. Citation2015). However, the least-processed BrC sampled in the ambient measurements corresponded to a transport time of ∼1 h. By this point, the changes to absorption and AAE resulting from ozonolysis would have already plateaued (), and thus our ozonolysis results and the field measurements are not directly comparable.
Despite the rapid kinetics of heterogeneous ozonolysis, BrC will continue to bleach over longer time periods due to ongoing heterogeneous oxidation by OH. The lifetime of BrC with respect to heterogeneous oxidation by OH can be calculated by determining an effective uptake coefficient (), the fraction of OH-condensed phase molecule collisions resulting in reaction. As the molecules contributing to absorption are unknown and may continue to absorb after one generation of oxidation, there are some challenges in applying this analysis to aerosol absorption. However, if we approximate the aerosol as composed of one generic, weakly absorbing organic compound that decays at the same rate as k405 and has the average molecular weight of the measured aerosol components, we can estimate an uptake coefficient for k405. This definition of
is independent of the extent of reaction and can be calculated using the following equation:
(2)
(2)
where
is the surface-area-weighted mean particle diameter,
is the density of the particle (assumed 1.3 g cm−3),
is Avagadro’s number,
is the second-order rate constant calculated in Section 3.2,
is the mean speed of the OH molecules (619 m s−1), and M is the average molecular weight. We estimate the average molecular weight as 202 g/mol (the average molecular weight for the mass spectra acquired at the lowest OH exposure). This results in an approximate
of ∼0.5. Given the numerous uncertainties in this calculation, we do not correct for diffusion. Assuming a 200 nm particle and average OH concentration of 1.5 × 106 molecules cm−3, this uptake coefficient corresponds to a lifetime of BrC with respect to oxidation by OH of ∼13 days. There are uncertainties associated with this
and lifetime calculation given the limited number of experiments as well as the approximations and assumptions made in treating the aerosol as composed of a generic weakly absorbing compound. However, we can place an upper limit on
as the measurements are inconsistent with
(lifetime of ≤6 days for a 200 nm particle). As a typical atmospheric lifetime of submicron aerosol is ∼1–2 weeks, this timescale, even with the associated uncertainty, suggests that BrC will continue to be modified over its entire atmospheric lifetime.
To our knowledge, only two other studies have investigated the heterogeneous oxidation of primary biomass burning BrC (Li et al. Citation2019; Sumlin et al. Citation2017). Both found a decrease in absorption and increase in oxygen content of the aerosols with aging on a timescale of days. Although ozone was present in both these experiments as well, the role of aging by ozone alone was investigated only by Li et al. (Citation2019). Although the results of Li et al. (Citation2019) hinted at bleaching by ozone, the uncertainty in the k was too large for definitive conclusions.
The kinetics of bleaching differ among the two previous studies and the present one. Sumlin et al. (Citation2017) observed bleaching only after 3 days of aging, whereas Li et al. (Citation2019) observed fairly rapid initial bleaching over the first few days followed by slower bleaching. In our measurements, continual changes to the aerosol optical properties over all ages studied were observed. The changes were driven initially by ozone for atmospheric lifetimes of minutes to hours and then by OH for lifetimes of days. Continual aging of the absorption is expected given the continuous evolution of chemical composition. The reason for the difference between the various studies is not completely clear, and likely results from differences in the BrC composition/generation. Additionally, the timescales may not be directly comparable between our work and the Sumlin et al. (Citation2017) study, as in the work by Sumlin et al. (Citation2017), OH exposures (and hence equivalent atmospheric ages) were only estimated. The suppression of OH by any smoldering-derived VOCs that made it through the charcoal denuder and into the oxidation reactor would not be included in such estimates, and hence the OH levels and equivalent ages would be overestimated, particularly at early low oxidation levels. In this work, OH exposure was measured by a tracer technique, so these effects were included in the estimate of atmospheric aging time.
Despite these differences, the results from the three laboratory studies are broadly consistent, and moreover qualitatively agree with ambient measurements of BrC aging, which find that BrC becomes less absorbing with atmospheric age (Forrister et al. Citation2015; Wang et al. Citation2018). Additionally, ambient measurements find that AAE decreases with age, also consistent with our OH aging experiment (). The kinetics of this aging process appear to be different between the laboratory and ambient measurements: the sustained bleaching timescale of BrC observed here and by previous laboratory experiments (Li et al. Citation2019; Sumlin et al. Citation2017) is much longer than the shorter bleaching timescales (∼1 day) observed by ambient measurements. The difference between the laboratory and ambient measurements may be attributable to the difficulties in constraining the evolution of the optical properties of ambient, primary BrC at timescales longer than 1–2 days. At these longer timescales, processes such as dilution and OA formation complicate data interpretation, particularly if there is secondary BrC formation. A recent study investigated the heterogeneous oxidation of secondary BrC and found that at low RH (15%), absorption steadily increased while at higher RH (60%), the initial absorption enhancement was followed by rapid bleaching (Schnitzler and Abbatt Citation2018). Overall, it seems likely that a wide range of aging processes including heterogeneous oxidation, photolysis and photobleaching, SOA formation, aqueous processing, and evaporation act to alter BrC optical properties in the atmosphere. Given the complexity of this process and the limited number of systems studied both in the laboratory and the field, the differences between the studies are perhaps unsurprising and indicate that further investigation is required.
4. Conclusions
The work presented here suggests that under the set of conditions investigated, heterogeneous oxidation decreases the extent to which BrC particles absorb light. For the aerosol generated in this work, bleaching initially proceeds quickly due to reaction with ozone; however, a fraction of the absorbing compounds may be either nonreactive with or inaccessible to ozone. Continued bleaching occurs with a lifetime of ∼2 weeks for a 200 nm particle due to heterogeneous oxidation by OH. The timescale of this continued evolution contrasts with results suggesting that bleaching occurs on the timescale of days (Forrister et al. Citation2015; Wang et al. Citation2018) but agrees with other results showing sustained oxidation due to heterogeneous oxidation by OH (Sumlin et al. Citation2017).
Overall, BrC is undoubtedly a dynamic system, which ages by complex and variable mechanisms. The limited number of cases investigated here cannot fully capture the complexity of BrC aging. Continued investigation of BrC heterogeneous oxidation kinetics is required to understand if heterogeneous oxidation affects BrC evolution more generally. As the ability of models to accurately estimate the radiative effects of BrC depend on assumptions regarding BrC lifetime, continued investigation of the kinetics of BrC evolution from all aging mechanisms is required. This includes both additional laboratory studies (investigating how various aging processes affect the properties of a wide range of biomass burning aerosol types) and more ambient studies of changes to BrC with atmospheric age, ideally extending beyond lifetimes of only 1–2 days (which can be extremely challenging given the extent of dilution over these longer times). Such measurements will enable the direct comparison between process-based studies and ambient observations, providing insight into the factors governing light absorption by BrC, and ultimately the impacts of biomass burning emissions on present and future climate.
Supplemental Material
Download Zip (671 KB)Additional information
Funding
Related Research Data
References
- Adler, G., J. M. Flores, A. Abo Riziq, S. Borrmann, and Y. Rudich. 2011. Chemical, physical, and optical evolution of biomass burning aerosols: A case study. Atmos. Chem. Phys. 11 (4):1491–1503. doi: 10.5194/acp-11-1491-2011.
- Alfarra, M. R., A. S. H. Prevot, S. Szidat, J. Sandradewi, S. Weimer, V. A. Lanz, D. Schreiber, M. Mohr, and U. Baltensperger. 2007. Identification of the mass spectral signature of organic aerosols from wood burning emissions. Environ. Sci. Technol. 41 (16):5770–5777. doi: 10.1021/es062289b.
- Bluvshtein, N., P. Lin, J. M. Flores, L. Segev, Y. Mazar, E. Tas, G. Snider, C. Weagle, S. S. Brown, A. Laskin, and Y. Rudich. 2017. Broadband optical properties of biomass burning aerosol and identification of brown carbon chromophores. J. Geophys. Res. Atmos. 122 (10):5441–5456. doi: 10.1002/2016JD026230.
- Bond, T. C., D. G. Streets, K. F. Yarber, S. M. Nelson, J.-H. Woo, and Z. Klimont. 2004. A technology-based global inventory of black and organic carbon emissions from combustion. J. Geophys. Res. Atmos. 109 (D14):D14203. doi: 10.1029/2003JD003697.
- Cappa, C. D., D. L. Che, S. H. Kessler, J. H. Kroll, and K. R. Wilson. 2011. Variations in organic aerosol optical and hygroscopic properties upon heterogeneous OH oxidation. J. Geophys. Res. Atmos. 116 (D15):D15204. doi: 10.1029/2011JD015918.
- Cappa, C. D., T. B. Onasch, P. Massoli, D. R. Worsnop, T. S. Bates, E. S. Cross, P. Davidovits, J. Hakala, K. L. Hayden, B. T. Jobson, K. R. Kolesar, D. A. Lack, B. M. Lerner, S.-M. Li, D. Mellon, I. Nuaaman, J. S. Olfert, T. Petaja, P. K. Quinn, C. Song, R. Subramanian, E. J. Williams, and R. A. Zaveri. 2012. Radiative absorption enhancements due to the mixing state of atmospheric black carbon. Science 337 (6098):1078–1081. doi: 10.1126/science.1223447.
- Chakrabarty, R. K., H. Moosmüller, L.-W. A. Chen, K. Lewis, W. P. Arnott, C. Mazzoleni, M. K. Dubey, C. E. Wold, W. M. Hao, and S. M. Kreidenweis. 2010. Brown carbon in tar balls from smoldering biomass combustion. Atmos. Chem. Phys. 10 (13):6363–6370. doi: 10.5194/acp-10-6363-2010.
- Chang, J. L., and J. E. Thompson. 2010. Characterization of colored products formed during irradiation of aqueous solutions containing H2O2 and phenolic compounds. Atmos. Environ. 44 (4):541–551. doi: 10.1016/j.atmosenv.2009.10.042.
- Chen, Y., and T. C. Bond. 2010. Light absorption by organic carbon from wood combustion. Atmos. Chem. Phys. 10 (4):1773–1787. doi: 10.5194/acp-10-1773-2010.
- Chung, C. E., V. Ramanathan, and D. Decremer. 2012. Observationally constrained estimates of carbonaceous aerosol radiative forcing. Proc. Natl. Acad. Sci. 109 (29):11624–11629. doi: 10.1073/pnas.1203707109.
- DeCarlo, P. F., J. R. Kimmel, A. Trimborn, M. J. Northway, J. T. Jayne, A. C. Aiken, M. Gonin, K. Fuhrer, T. Horvath, K. S. Docherty, D. R. Worsnop, and J. L. Jimenez. 2006. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 78 (24):8281–8289. doi: 10.1021/ac061249n.
- Feng, Y., V. Ramanathan, and V. R. Kotamarthi. 2013. Brown carbon: A significant atmospheric absorber of solar radiation? Atmos. Chem. Phys. 13 (17):8607–8621. doi: 10.5194/acp-13-8607-2013.
- Forrister, H., J. Liu, E. Scheuer, J. Dibb, L. Ziemba, K. L. Thornhill, B. Anderson, G. Diskin, A. E. Perring, J. P. Schwarz, P. Campuzano-Jost, D. A. Day, B. B. Palm, J. L. Jimenez, A. Nenes, and R. J. Weber. 2015. Evolution of brown carbon in wildfire plumes. Geophys. Res. Lett. 42 (11):4623–4630. doi: 10.1002/2015GL063897.
- Gelencsér, A., A. Hoffer, G. Kiss, E. Tombácz, R. Kurdi, and L. Bencze. 2003. In-situ formation of light-absorbing organic matter in cloud water. J. Atmos. Chem. 45 (1):25–33. doi: 10.1023/A:1024060428172.
- George, I. J., and J. P. D. Abbatt. 2010. Heterogeneous oxidation of atmospheric aerosol particles by gas-phase radicals. Nat. Chem. 2 (9):713–722. doi: 10.1038/nchem.806.
- Hallquist, M., J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prévôt, J. H. Seinfeld, J. D. Surratt, R. Szmigielski, and J. Wildt. 2009. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos. Chem. Phys. 9 (14):5155–5236. doi: 10.5194/acp-9-5155-2009.
- Hems, R. F., and J. P. D. Abbatt. 2018. Aqueous phase photo-oxidation of brown carbon nitrophenols: Reaction kinetics, mechanism, and evolution of light absorption. ACS Earth Space Chem. 2 (3):225–234. doi: 10.1021/acsearthspacechem.7b00123.
- Jimenez, J. L., M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger, and D. R. Worsnop. 2009. Evolution of organic aerosols in the atmosphere. Science 326 (5959):1525–1529. doi: 10.1126/science.1180353.
- Jo, D. S., R. J. Park, S. Lee, S.-W. Kim, and X. Zhang. 2016. A global simulation of brown carbon: Implications for photochemistry and direct radiative effect. Atmos. Chem. Phys. 16 (5):3413–3432. doi: 10.5194/acp-16-3413-2016.
- Kroll, J. H., J. D. Smith, D. R. Worsnop, and K. R. Wilson. 2012. Characterisation of lightly oxidised organic aerosol formed from the photochemical aging of diesel exhaust particles. Environ. Chem. 9 (3):211–220. doi: 10.1071/EN11162.
- Lack, D. A., M. S. Richardson, D. Law, J. M. Langridge, C. D. Cappa, R. J. McLaughlin, and D. M. Murphy. 2012. Aircraft instrument for comprehensive characterization of aerosol optical properties, part 2: Black and brown carbon absorption and absorption enhancement measured with photo acoustic spectroscopy. Aerosol Sci. Technol. 46 (5):555–568. doi: 10.1080/02786826.2011.645955.
- Lambe, A. T., C. D. Cappa, P. Massoli, T. B. Onasch, S. D. Forestieri, A. T. Martin, M. J. Cummings, D. R. Croasdale, W. H. Brune, D. R. Worsnop., et al. 2013. Relationship between oxidation level and optical properties of secondary organic aerosol. Environ. Sci. Technol. 47 (12):6349–6357. doi: 10.1021/es401043j.
- Langridge, J. M., M. S. Richardson, D. Lack, D. Law, and D. M. Murphy. 2011. Aircraft instrument for comprehensive characterization of aerosol optical properties, part I: Wavelength-dependent optical extinction and its relative humidity dependence measured using cavity ringdown spectroscopy. Aerosol Sci. Technol. 45 (11):1305–1318. doi: 10.1080/02786826.2011.592745.
- Laskin, A., J. Laskin, and S. A. Nizkorodov. 2015. Chemistry of atmospheric brown carbon. Chem. Rev. 115 (10):4335–4382. doi: 10.1021/cr5006167.
- Li, C., Q. He, J. Schade, J. Passig, R. Zimmermann, D. Meidan, A. Laskin, and Y. Rudich. 2019. Dynamic changes in optical and chemical properties of tar ball aerosols by atmospheric photochemical aging. Atmos. Chem. Phys. 19 (1):139–163. doi: 10.5194/acp-19-139-2019.
- Lin, G., J. E. Penner, M. G. Flanner, S. Sillman, L. Xu, and C. Zhou. 2014. Radiative forcing of organic aerosol in the atmosphere and on snow: Effects of SOA and brown carbon. J. Geophys. Res. Atmos. 119 (12):7453–7476. doi: 10.1002/2013JD021186.
- Lin, P., N. Bluvshtein, Y. Rudich, S. A. Nizkorodov, J. Laskin, and A. Laskin. 2017. Molecular chemistry of atmospheric brown carbon inferred from a nationwide biomass burning event. Environ. Sci. Technol. 51 (20):11561–11570. doi: 10.1021/acs.est.7b02276.
- Lin, P., L. T. Fleming, S. A. Nizkorodov, J. Laskin, and A. Laskin. 2018. Comprehensive molecular characterization of atmospheric brown carbon by high resolution mass spectrometry with electrospray and atmospheric pressure photoionization. Anal. Chem. 90 (21):12493–12502. doi: 10.1021/acs.analchem.8b02177.
- Malecha, K. T., Z. Cai, and S. A. Nizkorodov. 2018. Photodegradation of secondary organic aerosol material quantified with a quartz crystal microbalance. Environ. Sci. Technol. Lett. 5 (6):366–371. doi: 10.1021/acs.estlett.8b00231.
- Mohr, C., F. D. Lopez-Hilfiker, P. Zotter, A. S. H. Prévôt, L. Xu, N. L. Ng, S. C. Herndon, L. R. Williams, J. P. Franklin, M. S. Zahniser, D. R. Worsnop, W. B. Knighton, A. C. Aiken, K. J. Gorkowski, M. K. Dubey, J. D. Allan, J. A. Thornton. 2013. Contribution of nitrated phenols to wood burning brown carbon light absorption in Detling, United Kingdom during winter time. Environ. Sci. Technol. 47 (12):6316–6324. doi: 10.1021/es400683v.
- Northway, M. J., J. T. Jayne, D. W. Toohey, M. R. Canagaratna, A. Trimborn, K. I. Akiyama, A. Shimono, J. L. Jimenez, P. F. DeCarlo, K. R. Wilson, and D. R. Worsnop. 2007. Demonstration of a VUV lamp photoionization source for improved organic speciation in an aerosol mass spectrometer. Aerosol Sci. Technol. 41 (9):828–839. doi: 10.1080/02786820701496587.
- Oros, D. R., and B. R. T. Simoneit. 2001. Identification and emission factors of molecular tracers in organic aerosols from biomass burning part 1. Temperate climate conifers. Appl. Geochem. 16 (13):1513–1544. doi: 10.1016/S0883-2927(01)00021-X.
- Ortega, A. M., Day, D. A. M. J. Cubison, W. H. Brune, D. Bon, J. A. de Gouw, and J. L. Jimenez. 2013. Secondary organic aerosol formation and primary organic aerosol oxidation from biomass-burning smoke in a flow reactor during FLAME-3. Atmos. Chem. Phys. 13 (22):11551–11571. doi: 10.5194/acp-13-11551-2013.
- Pechony, O., and D. T. Shindell. 2010. Driving forces of global wildfires over the past millennium and the forthcoming century. Proc. Natl. Acad. Sci. 107 (45):19167–19170. doi: 10.1073/pnas.1003669107.
- Saleh, R., C. J. Hennigan, G. R. McMeeking, W. K. Chuang, E. S. Robinson, H. Coe, N. M. Donahue, and A. L. Robinson. 2013. Absorptivity of brown carbon in fresh and photo-chemically aged biomass-burning emissions. Atmos. Chem. Phys. 13 (15):7683–7693. doi: 10.5194/acp-13-7683-2013.
- Saleh, R., M. Marks, J. Heo, P. J. Adams, N. M. Donahue, and A. L. Robinson. 2015. Contribution of brown carbon and lensing to the direct radiative effect of carbonaceous aerosols from biomass and biofuel burning emissions. J. Geophys. Res. Atmos. 120 (19):10285–10296. doi: 10.1002/2015JD023697.
- Saleh, R., E. S. Robinson, D. S. Tkacik, A. T. Ahern, S. Liu, A. C. Aiken, R. C. Sullivan, A. A. Presto, M. K. Dubey, R. J. Yokelson, N. M. Donahue, and A. L. Robinson. 2014. Brownness of organics in aerosols from biomass burning linked to their black carbon content. Nat. Geosci. 7 (9):647–650. doi: 10.1038/ngeo2220.
- Schneider, J., S. Weimer, F. Drewnick, S. Borrmann, G. Helas, P. Gwaze, O. Schmid, M. O. Andreae, and U. Kirchner. 2006. Mass spectrometric analysis and aerodynamic properties of various types of combustion-related aerosol particles. Int. J. Mass Spectrometry 258 (1–3):37–49. doi: 10.1016/j.ijms.2006.07.008.
- Schnitzler, E. G., and J. P. D. Abbatt. 2018. Heterogeneous OH oxidation of secondary brown carbon aerosol. Atmos. Chem. Phys. 18 (19):14539–14553. doi: 10.5194/acp-18-14539-2018.
- Seinfeld, J. H., and S. N. Pandis. 2006. Atmospheric chemistry and physics: from air pollution to climate change. 2nd ed. Hoboken, NJ: Wiley.
- Smith, J. D., J. H. Kroll, C. D. Cappa, D. L. Che, C. L. Liu, M. Ahmed, S. R. Leone, D. R. Worsnop, and K. R. Wilson. 2009. The heterogeneous reaction of hydroxyl radicals with sub-micron squalane particles: A model system for understanding the oxidative aging of ambient aerosols. Atmos. Chem. Phys. 9 (9):3209–3222. doi: 10.5194/acp-9-3209-2009.
- Stocker, T. F., D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex, and P. M. Midgley, eds. 2013. IPCC, 2013: Climate Change 2013: The physical science basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge and New York, NY: Cambridge University Press.
- Sumlin, B. J., A. Pandey, M. J. Walker, R. S. Pattison, B. J. Williams, and R. K. Chakrabarty. 2017. Atmospheric photooxidation diminishes light absorption by primary brown carbon aerosol from biomass burning. Environ. Sci. Technol. Lett. 4 (12):540–545. doi: 10.1021/acs.estlett.7b00393.
- Wang, X., C. L. Heald, D. A. Ridley, J. P. Schwarz, J. R. Spackman, A. E. Perring, H. Coe, D. Liu, and D. A. D. Clarke. 2014. Exploiting simultaneous observational constraints on mass and absorption to estimate the global direct radiative forcing of black carbon and brown carbon. Atmos. Chem. Phys. 14 (20):10989–11010. doi: 10.5194/acp-14-10989-2014.
- Wang, X., C. L. Heald, J. Liu, R. J. Weber, P. Campuzano-Jost, J. L. Jimenez, J. P. Schwarz, and A. E. Perring. 2018. Exploring the observational constraints on the simulation of brown carbon. Atmos. Chem. Phys. 18 (2):635–653. doi: 10.5194/acp-18-635-2018.
- Wong, J. P. S., A. Nenes, and R. J. Weber. 2017. Changes in light absorptivity of molecular weight separated brown carbon due to photolytic aging. Environ. Sci. Technol. 51 (15):8414–8421. doi: 10.1021/acs.est.7b01739.
- Zarzana, K. J., C. D. Cappa, and M. A. Tolbert. 2014. Sensitivity of aerosol refractive index retrievals using optical spectroscopy. Aerosol Sci. Technol. 48 (11):1133–1144. doi: 10.1080/02786826.2014.963498.
- Zhang, Q., J. L. Jimenez, M. R. Canagaratna, J. D. Allan, H. Coe, I. Ulbrich, M. R. Alfarra, A. Takami, A. M. Middlebrook, Y. L. Sun, K. Dzepina, E. Dunlea, K. Docherty, P. F. DeCarlo, D. Salcedo, T. Onasch, J. T. Jayne, T. Miyoshi, A. Shimono, S. Hatakeyama, N. Takegawa, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, P. Williams, K. Bower, R. Bahreini, L. Cottrell, R. J. Griffin, J. Rautiainen, J. Y. Sun, Y. M. Zhang, D. R. Worsnop. 2007. Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes. Geophys. Res. Lett. 34 (13):L13801. doi: 10.1029/2007GL029979
- Zhang, X., H. Kim, C. L. Parworth, D. E. Young, Q. Zhang, A. R. Metcalf, and C. D. Cappa. 2016. Optical properties of wintertime aerosols from residential wood burning in Fresno, CA: Results from DISCOVER-AQ 2013. Environ. Sci. Technol. 50 (4):1681–1690. doi: 10.1021/acs.est.5b04134.
- Zhao, R., A. K. Y. Lee, L. Huang, X. Li, F. Yang, and J. P. D. Abbatt. 2015. Photochemical processing of aqueous atmospheric brown carbon. Atmos. Chem. Phys. 15 (11):6087–6100. doi: 10.5194/acp-15-6087-2015.
- Zhong, M., and M. Jang. 2014. Dynamic light absorption of biomass-burning organic carbon photochemically aged under natural sunlight. Atmos. Chem. Phys. 14 (3):1517–1525. doi: 10.5194/acp-14-1517-2014.