
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Polycyclic aromatic hydrocarbons (PAHs) are important constituents of brown carbon (BrC) that are subject to atmospheric processing by gas-phase oxidants such as the hydroxyl radical (OH) and the nitrate radical (NO3). While OH oxidation of BrC has been investigated extensively, studies of NO3 oxidation are limited. Here, we generated BrC from the combustion of toluene containing a complex mixture of light-absorbing PAHs and investigated changes in their chemical composition and light-absorption properties following exposure to NO3 in an oxidation flow reactor. Three types of BrC were studied, with varying light-absorption properties that were classified in terms of the imaginary part of the refractive index (k) as: light BrC (k at 532 nm, k532 = 0.008), medium BrC (k532 = 0.026), and dark BrC (k532 = 0.091). Exposure to NO3 led to ∼30% increase in k532 of the light and medium BrC and ∼5% decrease in k532 of the dark BrC. This discrepancy is attributed to two competing effects: 1) addition of chromophoric functional groups by NO3-induced heterogeneous oxidation and 2) condensation of gas-phase PAH + NO3 oxidation products that were less-absorbing than the particulate PAHs. Analysis of the aerosol chemical evolution revealed that effect (2) was more important in the dark BrC experiments. We performed optical calculations to isolate effect (1) and showed that heterogeneous oxidation led to ∼50% increase in k532 for all the BrC types. These results indicate that NO3-induced heterogeneous oxidation darkens some types of atmospheric BrC, which can counterbalance bleaching effects induced by OH oxidation.
Copyright © 2020 American Association for Aerosol Research
EDITOR:
1. Introduction
Brown carbon (BrC), also known as light-absorbing organic aerosol (OA), plays a vital role alongside black carbon (BC) in the absorption of solar radiation by carbonaceous aerosols. This has been confirmed by numerous laboratory experiments (e.g., Saleh et al. Citation2014; Adler et al. Citation2019; Olson et al. Citation2015), field observations (e.g., Lack et al. Citation2012; Zhang et al. Citation2017), and remote-sensing observations (e.g., Wang et al. Citation2016; Jethva and Torres Citation2011; Wang et al. Citation2013). Consequently, the radiative effect of light absorption by BrC has been the focus of several global-climate calculations (e.g., Saleh et al. Citation2015; Jo et al. Citation2016; Hammer et al. Citation2016; Wang et al. Citation2014; Brown et al. Citation2018; Feng, Ramanathan, and Kotamarthi Citation2013; Wang et al. Citation2018). These modeling studies have revealed a potentially important, yet highly uncertain radiative effect associated with BrC light absorption, with global-mean values ranging between +0.03 W m−2 and +0.57 W m−2. This large variability is in part a manifestation of the limited understanding of the chemical nature, formation pathways, and atmospheric processing of BrC, which hinders explicit representation of the wide range of experimentally derived BrC light-absorption properties within a climate-modeling framework.
While BrC is typically associated with biomass-combustion emissions, it has also been observed in the emissions of fossil-fuel combustion as well as in secondary organic aerosol (SOA) formed by the oxidation of biogenic and anthropogenic precursors (Laskin, Laskin, and Nizkorodov Citation2015; Moise, Flores, and Rudich 2015). The light-absorption properties of BrC are usually quantified using the wavelength-dependent imaginary part of the refractive index (k) or the mass absorption cross section (MAC). The values of k at mid-visible wavelengths reported for BrC range over 3 orders of magnitude (10−4 to 10−1) (Laskin, Laskin, and Nizkorodov Citation2015). BrC exhibits an increase in absorption with decreasing wavelength within the visible spectrum, which can be represented using a power-law functional dependence on wavelength. The values of wavelength dependence (w) of k (i.e., the exponent of the power-law) vary from ∼1 to 10. For reference, the corresponding mid-visible MAC (m2 g−1) values can be roughly approximated by multiplying the mid-visible k by 10. Also, the wavelength dependence of MAC, the absorption Ångström exponent (AAE), can be approximated as w + 1 for particles much smaller than the wavelength.
In terms of chemical composition, atmospheric BrC has been often associated with the broad family of humic-like substances (HULIS) (Kwon et al. Citation2018; Claeys et al. Citation2012; Utry et al. Citation2013; Wang et al. Citation2019). This is rooted in a study by Mukai and Ambe (Citation1986) who identified brown airborne particles originating from biomass burning and found that their chemical signatures were similar to humic acids. Another broad term, “tar balls,” was introduced by Posfai et al. (Citation2004) and has been used to designate a class of BrC emitted from biomass combustion (Adachi et al. Citation2018; Sedlacek III et al. 2018; Hoffer et al. Citation2016; Tóth et al. Citation2014). These two classifications are not necessarily incompatible. BrC absorption in biomass-combustion emissions have been linked to charge transfer complexes (Phillips and Smith Citation2014) as well as aromatic species, including polycyclic aromatic hydrocarbons (PAHs) (Li, He, Schade et al. Citation2019; Adler et al. Citation2019) and oxygenated and nitrated aromatics (Li, He, Schade et al. Citation2019; Lin et al. Citation2016; Liu et al. Citation2017; Desyaterik et al. Citation2013). Such chromophores have been identified both in HULIS (Claeys et al. Citation2012; Wang et al. Citation2019) as well as tar balls (Li, He, Hettiyadura et al. Citation2019; Li, He, Schade et al. Citation2019).
Despite the advances in identifying the chemical species that constitute BrC, a comprehensive picture of the landscape of BrC chemical structure is still lacking (Laskin, Laskin, and Nizkorodov Citation2015). Perhaps even more lacking is the understanding of the evolution of BrC upon chemical processing in the atmosphere, which has been the focus of numerous recent studies. Both laboratory studies (Browne et al. Citation2019; Sumlin et al. Citation2017; Zhao et al. Citation2015; Wong, Nenes, and Weber Citation2017; Wong et al. Citation2019) and field measurements (Satish et al. Citation2017; Dasari et al. Citation2019; Forrister et al. Citation2015; Wong et al. Citation2019) have observed that photo-oxidation led to a reduction in BrC light absorption. This bleaching effect could be attributed to either dark or photo-initiated destruction of chromophores through OH oxidation. On the other hand, the presence of NOx has been observed to enhance the BrC light absorption (Li, He, Hettiyadura et al. Citation2019; Li, He, Schade et al. Citation2019; Wang et al. Citation2019) or slow down the OH-induced bleaching effect (Zhong and Jang Citation2014; Nakayama et al. Citation2013). Another important, yet understudied, atmospheric reaction that can potentially contribute to the dynamic behavior of BrC light absorption is the nighttime reaction of PAHs with NO3 radicals (Keyte, Harrison, and Lammel Citation2013;). The large reaction rate coefficients (Mak, Gross, and Bertram Citation2007) and rapid uptake of NO3 on surfaces (Knopf, Forrester, and Slade Citation2011) suggest that NO3 could induce heterogeneous oxidation of condensed-phase PAHs, leading to the formation of nitrogen-containing PAH derivatives, including nitro-PAHs. Studies of heterogeneous oxidation of single-compound PAHs (e.g., anthracene, pyrene, and benzo[a]pyrene) with NO2/NO3/N2O5 indicated that these reactions resulted in enhanced UV-visible absorption of the oxidized PAHs relative to their parent molecules (Kwamena and Abbatt Citation2008; Lu et al. Citation2011).
As alluded to above, PAHs are major constituents of combustion BrC, especially the highly absorptive fraction (Saleh, Cheng, and Atwi Citation2018; Li, He, Hettiyadura et al. Citation2019; Li, He, Schade et al. Citation2019; Adler et al. Citation2019). This is not surprising since PAHs have been identified as key species in the soot-formation process by numerous fundamental laboratory studies (Michelsen Citation2017) and have been known to constitute a substantial fraction of combustion emissions of fossil (Karavalakis et al. Citation2011) and biomass (Shen et al. Citation2012) fuels. We have recently shown that BrC produced from the combustion of benzene and toluene has prominent PAH signatures and exhibits varying molecular sizes, volatilities, and light-absorbing properties that are dependent on combustion conditions such as temperature and relative amounts of fuel, oxygen, and nitrogen (Saleh, Cheng, and Atwi Citation2018). The light-absorption properties of these PAHs fall on a continuum that extends from weakly absorbing (light) BrC to strongly absorbing (dark) BrC, eventually approaching BC (Cheng et al. Citation2019). Here, we build on these findings to investigate the evolution of the light-absorption properties of complex PAH-containing BrC following oxidation by NO3. We performed toluene-combustion experiments controlled at different combustion conditions to produce BrC with varying levels of darkness. For each combustion condition, we exposed the BrC to NO3 in an oxidation flow reactor (OFR) and compared the light-absorption properties and chemical composition of the oxidation products to their parent molecules. Since PAHs and their derivatives are the dominant constituents of toluene-combustion BrC (Saleh, Cheng, and Atwi Citation2018), we use BrC and PAHs interchangeably throughout the article in a manner that serves the discussion.
2. Methods
As shown in , the experimental setup employed in this study has 4 major components: 1) A combustion system used to generate BrC with variable light-absorption properties; 2) N2O5 generation system; 3) oxidation flow reactor (OFR), where the BrC was aged with NO3 produced from thermal decomposition of N2O5; and 4) a suite of instruments that enabled characterization of the chemical composition and light-absorption properties of the OFR effluent. Each of the components is described in the subsequent sections.
Figure 1. Schematic of the experimental setup for generating unoxidized and oxidized (with NO3) BrC.
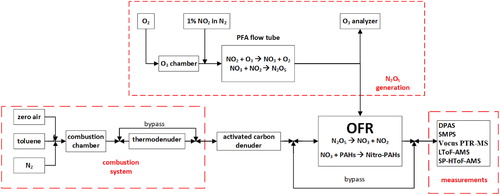
2.1. Combustion system
A detailed description of the combustion system can be found in Cheng et al. (Citation2019). Briefly, combustion of toluene was performed in a quartz combustion chamber controlled at 1050 °C. Toluene vapor was introduced into the combustion chamber by flowing nitrogen through a bubbler containing toluene. The flow of toluene-saturated nitrogen (0.14 − 0.17 LPM) was then mixed with flows of nitrogen (0.35 − 0.41 LPM) and zero air (0.23 − 0.24 LPM) before entering the combustion chamber. By controlling the relative flowrates of toluene, zero air, and nitrogen, we generated BrC with varying light-absorption properties. Specifically, decreasing the nitrogen flowrate produced larger-molecular size, more-absorbing (darker) BrC. Combustion conditions were fine-tuned using the observed light absorption properties (MAC and AAE) that were calculated in real-time based on measurements of particle size distributions and light absorption coefficients, as described in Section 2.3. In one experiment, we passed the combustion emissions through a thermodenuder controlled at 200 °C. By doing so, we relied on the established association between volatility, molecular size, and light-absorption properties (Saleh, Cheng, and Atwi Citation2018) to isolate the dark, large-molecular-size fraction of the BrC. In order to minimize contributions of gas-phase PAH + NO3 oxidation products on the optical properties of the BrC (i.e., condensed-phase PAHs), an activated carbon denuder was placed in-line before the OFR to absorb gas-phase components of the combustion emissions.
2.2. NO3 generation and oxidative aging of the BrC
As shown in , separate flows containing NO2 (1% in N2, Praxair) and O3 were input to a perfluoro alkoxy (PFA) flow tube with 1" outer diameter and 60” length that was operated as a laminar flow reactor (LFR), where, upon mixing, N2O5 was generated in the gas phase from the reaction NO2 + O3 → NO3 + O2 followed by the reaction NO3 + NO2 → N2O5 (Lambe et al. Citation2020). In these experiments, the NO2 in the N2 flow rate was set between 0 and 15 cm3 min−1, and O3 was generated by passing 2 LPM of O2 through an ozone chamber housing a mercury fluorescent lamp (GPH212T5VH, Light Sources, Inc.). The output of the LFR was mixed with the combustion emissions and ∼12 LPM of dilution air prior to injection into an Aerodyne Potential Aerosol Mass oxidation flow reactor (OFR) (Lambe et al. Citation2011). The mean residence time in the OFR was approximately 70 s. The N2O5 generated in the LFR thermally decomposed at room temperature inside the OFR to generate NO3, which initiated oxidation of the PAHs (both in the condensed phase and vapor phase) introduced from the combustion system. The integrated NO3 exposure, that is, the product of the NO3 concentration and mean OFR residence time, was estimated based on results from a separate set of experiments as 5.2 × 1013 molec cm−3 s, corresponding to an equivalent atmospheric aging time of approximately 2.4 nights assuming a 12-h average ambient NO3 mixing ratio of 20 ppt (Atkinson Citation1991) and negligible daytime NO3 concentration. The calculations are shown in the online supplementary information (SI).
2.3. Size distributions and light-absorption properties
The size distributions of the unoxidized and oxidized BrC particles were measured using a scanning mobility particle sizer (SMPS, TSI). As shown in Figure S1 (SI), the volume distributions of the particles were scanned from 10 nm to 400 nm, with mode diameters between 60 nm and 80 nm. We also integrated the SMPS volume distributions and calculated the total aerosol mass concentrations (mp) using a density of 1.2 g cm−3. The density was obtained in a previous experiment on similar aerosol emissions using the tandem differential mobility analyzer – aerosol particle mass analyzer (tandem DMA-APM) technique (Malloy et al. Citation2009).
We measured the absorption coefficients (babs, Mm−1) at λ = 473, 532, and 671 nm using a differential photoacoustic aerosol spectrometer (DPAS, Aerodyne Research Inc.) (Yu et al. Citation2019). The DPAS contains two photoacoustic cells, where one cell measures light absorption by the total aerosol and gaseous sample, and the other cell measures the light absorption by the gaseous sample only. Aerosol light absorption is calculated as the difference in light absorption measured by the two cells. The DPAS has a time resolution of 10 s and detection limits of 0.20 Mm−1 at 671 nm, 0.22 Mm−1 at 532 nm, and 0.90 Mm−1 at 473 nm (Yu et al. Citation2019). Following the procedure of Yu et al. (Citation2019), we calibrated the DPAS absorption at the 3 wavelengths using synthetic pigment black particles (Cab-O-Jet 200 from Cabot Corp., Boston, MA, USA). Cab-O-Jet has spectroscopic and morphological properties similar to aged BC (MAC at 550 nm = 7.89 ± 0.25 m2 g−1 and AAE = 1.03 ± 0.09) (You et al. Citation2016). The calibration process involved size-selecting Cab-O-Jet particles of 300 nm electrical-mobility diameter using a differential mobility analyzer (DMA, TSI) at different concentrations and using linear regression to obtain the calibration coefficient at each wavelength as the slope of the DPAS microphone signal versus concentration.
Using the babs values from the DPAS and mp values from the SMPS, we calculated MAC and AAE of the unoxidized aerosol during the experiments as:
(1)
(1)
(2)
(2)
where MAC532 is the MAC at 532 nm, λ is the wavelength, and AAE was obtained by fitting a power-law function to EquationEquation (2)
(2)
(2) .
We used these real-time calculations to tune the combustion conditions to produce BrC with a range of light-absorption properties. Producing the dark BrC also involved passing the emissions through a thermodenuder maintained at 200 °C (see Section 2.1).
We retrieved the wavelength-dependent BrC imaginary part of the refractive indices (k) from these measurements using optical closure (Saleh et al. Citation2014; Lack et al. Citation2012). The optical closure procedure involved fitting babs at 473, 532, and 671 nm obtained using Mie theory calculations to babs measured by the DPAS. Inputs to Mie calculations included the particle size distributions measured by the SMPS and the real part of the refractive index. The latter was assumed to be wavelength-independent with a value between 1.6 and 1.7, which are typical values observed for BrC (Saleh et al. Citation2014; Li, He, Hettiyadura et al. Citation2019; Li, He, Schade et al. Citation2019). The imaginary part of the refractive indices at each wavelength (k473, k532, k671) were fitting parameters. We then calculated the wavelength dependence (w) as the exponent of a power-law fit of the retrieved k473, k532, k671 versus wavelength.
2.4. Chemical speciation
Volatile organic compounds (VOCs) and oxygenated VOCs (OVOCs) were measured using a Vocus Proton Transfer Reaction Time-of-Flight Mass Spectrometer (PTR-MS, Tofwerk AG/Aerodyne Research, Inc.) (Krechmer et al. Citation2018) with H3O+ reagent ion. PTR-MS data were analyzed using the Tofware software package (Tofwerk AG, Aerodyne Research, Inc.) implemented in IGOR Pro (Wavemetrics, Inc.). Ensemble mass spectra of the particles and their aerodynamic size distributions were measured with two Aerodyne time-of-flight aerosol mass spectrometers (AMS). One AMS was operated with the standard tungsten vaporizer configuration to enable detection of non-refractory aerosol components with flash vaporization at 600 °C followed by electron impact ionization and time-of-flight mass spectrometry (e.g., DeCarlo et al. Citation2006). This AMS was equipped with a long high-resolution (resolving power up to ∼8,000 Δm/m) time-of-flight mass spectrometer L-ToF-AMS. The other AMS, a soot particle aerosol mass spectrometer (SP-AMS), was operated with a λ = 1064 nm laser vaporizer to enable detection of aerosol components that absorb at 1064 nm as well as species internally mixed with the absorbing aerosols (Onasch et al. Citation2012). The tungsten vaporizer was removed so that only absorbing particles were detected. This AMS used a high resolution TOFMS with resolving power up to ∼4,000 Δm/m. Elemental analysis yielding O/C, H/C, and N/C ratios was performed on AMS measurements using the SQUIRREL/PIKA software package implemented in IGOR Pro (Aiken, Decarlo, and Jimenez Citation2007; Canagaratna et al. Citation2015). To calculate N/C, we assumed that AMS signals at m/z = 30 (NO+) and m/z = 46 (NO2+) were associated with PAH + NO3 oxidation products (details in Section 3.2.1) and therefore were classified as organics in the software.
3. Results and discussion
3.1. Evolution of light-absorption properties
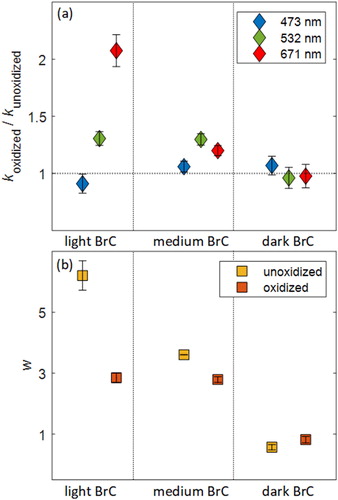
By tuning the combustion conditions of toluene, we generated BrC with varying light-absorption properties, which we categorized as light (k473 = 0.021, k532 = 0.008, k671 = 0.004, w = 6.2), medium (k473 = 0.053, k532 = 0.026, k671 = 0.020, w = 3.6), and dark (k473 = 0.099, k532 = 0.091, k671 = 0.081, w = 0.6). We note that the descriptors light, medium, and dark are not meant to be associated with the corresponding k values in an absolute sense, but are introduced to facilitate discussing the results. The evolution of the retrieved k and w after exposure to NO3 is depicted in , respectively. The k values of the oxidized BrC are normalized to those of the unoxidized BrC for ease of visual comparison across different wavelengths and different samples. The absolute k values of the oxidized BrC can be calculated by multiplying the ratios in with the values of unoxidized BrC above, and they are also given in Table S1 in the SI. The light BrC underwent a 10% decrease in k473, 30% increase in k532, and more than a factor of 2 increase in k671, which is manifested as an overall decrease in w of approximately 50% (). This indicates that NO3-induced oxidation added chromophores that are significantly darker (larger mid-visible k with a flatter wavelength dependence) than those existing in the parent PAHs. The medium BrC underwent a 7% increase in k473, 30% increase in k532, and 20% increase in k671. This indicates that for the medium BrC, NO3-induced oxidation also added chromophores that are darker than the original parent PAHs. The dark BrC experienced the smallest change in light-absorption properties. It underwent a 7% increase k473, 3% decrease in k532, and 2% decrease k671, which is an indication of the addition of chromophores that are, on average, relatively lighter than the parent PAHs. In Section 3.2, we discuss the differences in the evolution of light-absorption properties between the light, medium, and dark BrC in the context of differences in their chemical evolution in the OFR.
Figure 2. (a) Evolution of the imaginary part of refractive index (k) at λ = 473, 532, and 671 nm of light, medium, and dark BrC, quantified as the ratio of k of the oxidized BrC (koxidized) to k of the unoxidized BrC (kunoxidized). (b) The corresponding evolution of the wavelength dependence (w). Smaller w is indicative of darker BrC. Error bars represent standard deviations over time for one experiment. Numerical values of kunoxidized, koxidized, and w are given in Table S1.
3.2. Evolution of aerosol chemical composition
3.2.1. AMS and SP-AMS measurements of unoxidized and oxidized BrC
As described in Section 2.4, we characterized the aerosol chemical composition using an AMS and an SP-AMS. The AMS detected non-refractory components of the aerosol that flash-vaporized at ∼600 °C, whereas the SP-AMS detected components that absorbed the 1064 nm laser as well as components internally mixed with these strong absorbers. compares the normalized AMS () and SP-AMS () spectra of the unoxidized light and dark BrC aerosols. There are two points to make. First, the dark BrC spectra detected by both the AMS and SP-AMS contain organics with larger m/z compared to the light BrC spectra. This is in agreement with our previous results showing an association between BrC darkness and molecular size (Saleh, Cheng, and Atwi Citation2018). Second, for the same BrC (i.e., light or dark), the SP-AMS spectra are skewed to larger m/z compared to the AMS spectra. This can be interpreted based on the association between BrC molecular size, volatility, and light absorption (Saleh, Cheng, and Atwi Citation2018), and how these properties affect the detection efficiency of the two instruments. Specifically, the small-molecular-size BrC components have moderate volatilities and can readily flash-vaporize at 600 °C and be detected by the AMS. However, they are weakly absorbing and therefore do not efficiently absorb the 1064 nm laser, leading to low detection efficiency by the SP-AMS. On the other hand, the large-molecular-size BrC components have extremely low volatilities and do not readily flash-vaporize at 600 °C, leading to low detection efficiency by the AMS. However, they are strongly absorbing and can therefore efficiently absorb the 1064 nm laser (along with BC), leading to higher detection efficiency by the SP-AMS. However, BC is likely a minor component of the particles that were generated, for two reasons. First, as shown in Figure S1, the SMPS volume distribution is unimodal with a relatively small mode diameter (∼60 nm). The presence of BC would manifest as a second larger mode. Second SEM images of particles formed at similar combustion conditions in a previous study revealed near-spherical particles, indicative of pure OA, where BC-containing particles exhibited irregular shapes (Saleh, Cheng, and Atwi Citation2018).
Figure 3. AMS and SP-AMS UMR spectra of organic aerosol components in unoxidized light BrC and dark BrC aerosols.
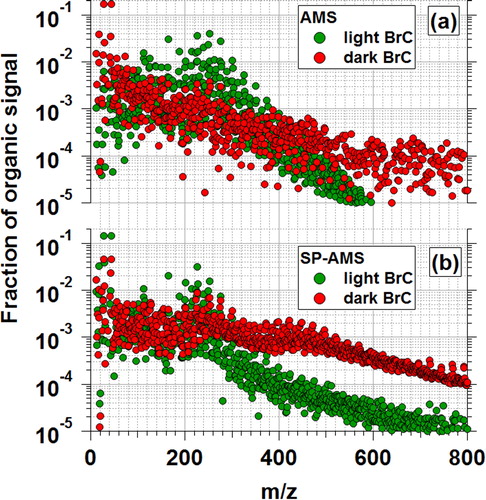
We observed a difference between the total OA concentrations detected by the AMS and SP-AMS for the light and dark BrC. For the light BrC, the OA concentration detected by the AMS was approximately a factor of 20 larger than the SP-AMS (Table S1). On the other hand, for the dark BrC the OA concentration detected by the SP-AMS was approximately a factor of 6 larger than the OA concentration detected by the AMS (Table S1). In addition to the large-molecular-size organic species shown in , the SP-AMS spectra of the dark BrC also contained signals at m/z = 12x that represent carbon clusters (Cx, where x = 1 to 9) typically associated with BC and a series of signals at m/z = 720 + 24x that represent trace fullerenes (C60 + 2x) present in the aerosols and/or formed in the laser (Onasch et al. 2015). These components likely also contributed to the absorption of the 1064 nm laser by the aerosol and overall increase in the SP-AMS detection efficiency.
This picture changed dramatically after oxidation. Due to the significant increase in light absorption induced by oxidation with NO3 (Section 3.1 and ), the oxidized light BrC was significantly more efficient at absorbing the SP-AMS 1064 nm laser than the parent molecules (i.e., unoxidized light BrC) resulting in more than an order-of-magnitude increase in detection efficiency by the SP-AMS. Consequently, the ratio of AMS OA to SP-AMS OA dropped from 20 to 2 after oxidation (Table S1). Results and discussion in the subsequent sections are based on AMS measurements of light and medium BrC and SP-AMS measurements of dark BrC.
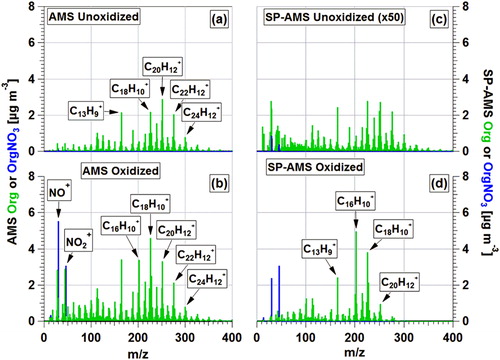
reveals additional noteworthy changes in the AMS and SP-AMS spectra of BrC following exposure to NO3 radicals in the OFR. First, NO3 exposure led to an increase in signals at m/z = 30 and 46 (NO+ and NO2+) and m/z = 44 (CO2+). Because negligible NH4+ is present, and because the NO+:NO2+ ratio in oxidized BrC (1.6-2.1) was higher than the NO+:NO2+ ratio in ammonium nitrate measured by the same AMS instrument (1.2), these signals can be attributed to PAH + NO3 oxidation products such as nitro-PAHs rather than particulate inorganic nitrates. However, we note that NO+:NO2+ values measured in this work are different from NO+:NO2+ of particulate organic nitrates studied previously (Bruns et al. Citation2010; Farmer et al. Citation2010), perhaps indicating different behavior of nitroaromatics in the AMS. The CO2+ signal is associated with decarboxylation of organic acids in the AMS (Canagaratna et al. Citation2015). Second, the organic mass per particle measured by the AMS increased following NO3 exposure, indicating OA enhancement by SOA formation or heterogeneous oxidation. Third, the oxidized BrC spectra () were shifted toward smaller carbon-number fragment ions than the corresponding unoxidized BrC spectra (). These changes may be due to condensation of gas-phase PAH + NO3 oxidation products and/or functionalization of condensed-phase PAHs following heterogeneous oxidation by NO3. The potential contributions of these processes are discussed in more detail in Section 3.2.3. Corresponding high-resolution AMS and SP-AMS spectra colored by CxHy, CxHyOz, CxHyOzN, CxHyN ion families are shown in Figures S3–S6 in the SI.
Figure 4. (a) AMS and (b) SP-AMS HR spectra of organic and nitrate signals in the unoxidized light BrC, and (c) AMS and (d) SP-AMS HR spectra of organic and nitrate signals in the oxidized light BrC (exposed to NO3 radicals at 2.4 nights of equivalent atmospheric NO3 exposure).
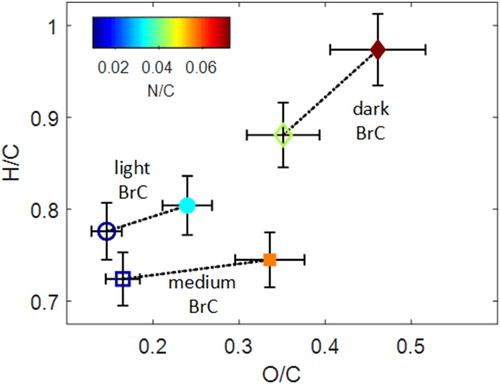
3.2.2. Evolution of elemental composition
The evolution of O/C, H/C, and N/C of the light, medium, and dark BrC due to oxidation with NO3 is depicted in the van Krevelen diagram (Heald et al. Citation2010) in . We calculated H/C and O/C ratio using the improved-ambient method (Canagaratna et al. Citation2015) and N/C ratio using the method of Aiken, Decarlo, and Jimenez (Citation2007). NO3 exposure resulted in an increase in H/C, O/C, and N/C for all PAHs. The increase in nitrogen content is associated with the addition of nitro (-NO2) groups (), either by heterogeneous oxidation or gas-phase oxidation followed by condensation. On the other hand, approximately half of the increase in oxygen content is associated with the addition of nitro groups and half was associated with the addition of other functional groups including carboxylic acids, carbonyls, and alcohols.
Figure 5. Van Krevelen diagram showing the H/C versus O/C for unoxidized (open symbols) and oxidized (solid symbols) BrC. Symbols are colored by N/C. Error bars represent ±12% uncertainty in O/C and ±4% uncertainty in H/C (Canagaratna et al. Citation2015).
3.2.3. SOA formation versus heterogeneous oxidation
The combustion emissions were passed through an activated carbon denuder () to remove residual toluene as well as gas-phase combustion products in order to minimize the effect of SOA formation relative to heterogeneous oxidation on the evolution of light-absorption properties of the BrC. The fractional removal of toluene and C10-C13 gas-phase PAHs was greater than 0.98, 0.99, 0.83, 0.92, and 0.13 for toluene, naphthalene (C10H8), acenaphthylene (C12H8), acenaphthene (C12H10), and fluorene (C13H10), respectively (Figure S2). Furthermore, after stripping semi-volatile PAHs from the gas phase, these species were expected to partition from the particle phase to the gas phase to maintain thermodynamic equilibrium, as appeared to be the case for phenanthrene/anthracene (C14H10), whose signal increased after denuding. Therefore, some SOA formation due to gas-phase NO3 oxidation and subsequent condensation of these semi-volatile PAHs is expected to occur.
To estimate the relative contribution of SOA formation versus heterogeneous oxidation to the increase in OA mass concentration after oxidation with NO3, we performed elemental (C, N, O) mass balance on the OA measured by the AMS and SP-AMS before and after oxidation. The functional groups that can potentially be added to the condensed phase by heterogeneous oxidation contain only nitrogen or oxygen. Therefore, any increase in carbon after oxidation was attributed to SOA formation. The carbon mass balance showed that SOA formation constituted 8%, 15%, and 37% of the carbon mass of the oxidized particles for the light, medium, and dark BrC, respectively. The relatively high SOA fraction in the dark BrC experiments may have been a consequence of semivolatile PAHs that evaporated inside the thermodenuder used to isolate the dark BrC (Section 2.1) and were subsequently oxidized in the OFR to generate SOA. As discussed in Section 3.3, the difference in the fractional contribution of SOA formation for the different BrC samples played a key role in the observed evolution in light-absorption properties ().
While the increase in carbon mass was clearly related to SOA formation, the increase in oxygen and nitrogen mass was potentially contributed from both SOA formation and/or heterogeneous oxidation processes. To estimate the relative contributions of each process, we applied the following constraints:
We assumed that condensable molecules formed from gas-phase PAH + NO3 reactions contained carbon numbers (C#) ranging from 10 (e.g., naphthalene) to 18 (e.g., chrysene). Known oxidation products of naphthalene (e.g., nitro-naphthalene) have saturation vapor pressures (Psat) > 10−2 Pa (Bandowe and Meusel Citation2017), corresponding to saturation concentration (msat) > 103 µg m−3, which is at the upper limit of what could significantly partition to the particle phase in our experiments, where typical OA mass concentrations (mOA) were 100-200 µg m−3. On the other hand, species with C# > 18 have Psat < 10−5 Pa (msat < 1 µg m−3) (Keyte, Harrison, and Lammel Citation2013) and are therefore expected to exist almost exclusively in the condensed phase.
We assumed that each condensing molecule contained nitrogen numbers (N#) ranging from 0 to 1 and oxygen numbers (O#) ranging from 2 to 4 (Sasaki et al. Citation1997; Zielinska et al. Citation1989; Atkinson et al. Citation1990). We did not impose upper limits on N# and O# contributed from heterogeneous oxidation processes (Liu et al. Citation2012).
Based on these constraints, we obtained possible ranges of the amounts of nitrogen and oxygen that can be brought into the particles via SOA formation (ΔNcond and ΔOcond) as a function of C# of the condensing molecules. We then subtracted these values from the total amounts of added nitrogen and oxygen (ΔNtot and ΔOtot) obtained from the mass balance to calculate the amounts of nitrogen and oxygen added via heterogeneous oxidation (ΔNhet and ΔOhet).
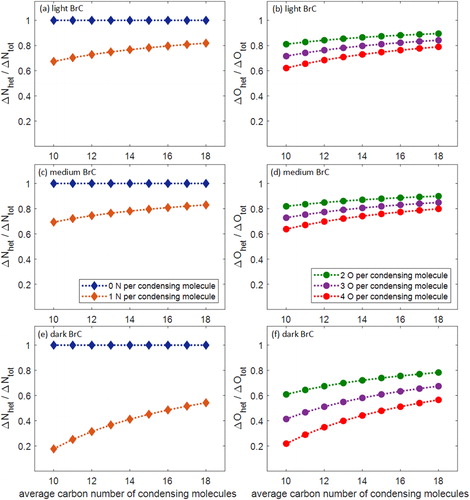
The results of these calculations are shown in for light, medium, and dark BrC. As expected, the contribution of heterogeneous oxidation to adding nitrogen (ΔNhet/ΔNtot) and oxygen (ΔOhet/ΔOtot) is inversely proportional to the assumed nitrogen and oxygen content of the SOA molecules. For the light and medium BrC, heterogeneous oxidation is expected to be the dominant mechanism in adding nitrogen and oxygen to the particles, with ΔNhet/ΔNtot ranging from 0.7 to 1 and ΔOhet/ΔOtot ranging from 0.6 to 0.9 as a function of C# for all assumed cases. On the other hand, the contribution of heterogeneous oxidation to dark BrC was more sensitive to the assumed composition of the condensing molecules, with ΔNhet/ΔNtot and ΔOhet/ΔOtot ranging from 0.2 to 1. While these calculations are speculative, they suggest that SOA formation may have a more prominent role in the chemical evolution of the dark BrC than for the light and medium BrC in our experiments, which is relevant to explaining the discrepancy in the evolution of light-absorption properties described in the next section.
Figure 6. Calculated relative contribution of heterogeneous oxidation to adding nitrogen (ΔNhet/ΔNtot) and oxygen (ΔOhet/ΔOtot) to the condensed phase as a function of assumed average carbon number of the condensing SOA molecules for (a, b) light, (c, d) medium, and (e, f) dark BrC.
3.3. The competing effects of SOA formation and heterogeneous oxidation on the evolution of light-absorption properties
As described in Section 3.1 and shown in , NO3-induced oxidation led to a relatively significant increase in darkness of the light BrC, a moderate increase in darkness of the medium BrC, and a minor decrease in darkness of the dark BrC. The different trends can be understood as:
The light-absorption properties of the oxidized BrC are average properties of the heterogeneously oxidized PAHs and the condensed oxidized PAHs (SOA).
The contribution of SOA to the average light-absorption properties is proportional to the relative amount of SOA, which is most significant for the dark BrC, followed by the medium BrC, then the light BrC (Section 3.2.3).
The molecular sizes of the gas-phase precursors of SOA are much smaller than the average molecular size of the unoxidized particulate PAHs and are therefore much less absorptive in the visible spectrum (Saleh, Cheng, and Atwi Citation2018). While the SOA generated from NO3 oxidation is more absorptive than its gas-phase precursors, it is expected to be significantly less absorptive than the unoxidized particulate PAHs if our N# ≤ 1 assumption is justified (Section 3.2.3). Therefore, SOA presumably decreases the overall absorption of oxidized BrC.
Heterogeneous oxidation with NO3 is expected to add chromophoric functional groups to the parent particulate PAHs (Lu et al. Citation2011; Zimmermann et al. Citation2013; Zhang et al. Citation2013; Jariyasopit et al. Citation2014) thus increasing the overall absorption by the oxidized BrC, especially if multiple nitrogen-containing functional groups are added (Liu et al. Citation2012).
To illustrate the competing effects of SOA formation and heterogeneous oxidation, we performed simplified calculations to isolate the effect of heterogeneous oxidation on the evolution of the light-absorption properties of the BrC particles. To do so, we represented the evolution of the BrC in the OFR in two phenomena: a heterogeneous oxidation step that changes the k of the BrC from kunoxidized to khet, and an SOA formation step that “dilutes” khet by adding the weakly absorbing SOA molecules to the BrC. To retrieve khet, we assumed that the k of the oxidized BrC (koxidized) is a mass-weighted average of khet and kSOA:
(3)
(3)
where ySOA and yhet are the corresponding mass fractions obtained from the mass balance (Section 3.2.3).
In EquationEquation (3)(3)
(3) , koxidized is retrieved from measurements of the oxidized BrC (), while kSOA is unknown but can be reasonably constrained. For the lower limit, we used kSOA,532 = 0.001, estimated from the data of Lee et al. (Citation2014) for SOA obtained from the high-NOx oxidation of naphthalene. For the upper limit, we used kSOA,532 = 0.08 which was obtained here for unoxidized light BrC, based on the assumption that SOA is less absorbing than unoxidized BrC. To extend these k-values to other wavelengths, we assumed w = 6.2, which is the value we obtained for the unoxidized light BrC. shows khet/kunoxidized for the range of assumed kSOA. For reference, we also show the data from , i.e., koxidized/kunoxidized, on the same plots.
Figure 7. Effect of heterogeneous oxidation on light-absorption properties of the light, medium, and dark BrC. The bars represent ranges of khet/kunoxidized calculated based on EquationEquation (3)(3)
(3) . They reflect the assumed range of kSOA as well as the uncertainty in koxidized and kunoxidized (Table S1). The diamonds represent koxidized/kunoxidized (the same as in ), added here for reference to compare the effect of heterogeneous oxidation to the overall observed oxidation. Error bars represent standard deviations over time for one.
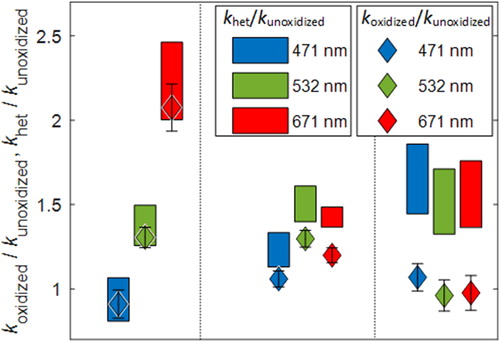
For the light BrC, SOA constituted only 8% of the carbon mass of the oxidized BrC. Therefore, addition of chromophoric functional groups via NO3-induced heterogeneous oxidation dominated the overall evolution of the light-absorption properties of the oxidized BrC because khet and koxidized were similar (). On the other hand, SOA constituted 37% of the carbon mass of the oxidized dark BrC and therefore had a more significant influence on its light-absorption properties. Assuming that the SOA was significantly less absorptive than the unoxidized particulate PAHs, it effectively “diluted” the overall absorption and led to the observed small decrease in absorption at the mid- and long-visible wavelengths ( and ). However, isolating the effect of heterogeneous oxidation reveals that, similar to the light BrC, reaction with NO3 also added chromophoric functional groups to the dark PAHs as manifested in an enhanced khet relative to kunoxidized ().
With the absence of knowledge of actual kSOA values, the calculations presented in are not meant to be quantitative. However, they indicate that heterogeneous oxidation with NO3 led to a similar increase in light absorption (i.e., similar khet/kunoxidized) for the light, medium, and dark BrC, and that the observed differences in the evolution of their light-absorption properties (koxidized/kunoxidized) may have been due to the difference in SOA contribution to the particle mass and overall absorption.
Our results are qualitatively consistent with the recent findings of Li, He, Hettiyadura et al. (Citation2019). That study reported an increase in k of wood-tar BrC of approximately a factor of 2 in the visible wavelengths and 40% in the UV wavelengths after 13.3 h of equivalent atmospheric NO3 oxidation in an OFR. The atmospheric-equivalent NO3 exposure in Li, He, Hettiyadura et al. (Citation2019) was shorter than our study (13.3 h vs 2.4 nights), yet their absorption enhancements are on the high end of ours. This could possibly be due to differences in experimental conditions or BrC chemical structure. Nevertheless, both studies indicate that heterogeneous NO3 oxidation is an important mechanism for enhancing BrC absorption that should be further investigated using more diverse BrC systems and NO3 exposure conditions.
3.4. Atmospheric implications
Our results suggest that light absorption by BrC is enhanced following NO3-induced heterogenous oxidation, while condensable products of gas-phase PAH + NO3 reactions may partially offset this enhancement. The majority of studies on atmospheric evolution of BrC have focused on daytime aging, leading to the large body of evidence on the bleaching of BrC due to oxidative destruction of BrC chromophores driven by OH oxidation or photolysis (e.g., Browne et al. Citation2019; Sumlin et al. Citation2017; Zhao et al. Citation2015; Wong, Nenes, and Weber Citation2017; Wong et al. Citation2019; Satish et al. Citation2017; Dasari et al. Citation2019; Forrister et al. Citation2015; Sarkar et al. Citation2019). On the other hand, observations of nighttime atmospheric evolution of BrC are scarce. Two complementary studies (Lin et al. Citation2017; Bluvshtein et al. Citation2017) performed optical and chemical speciation measurements of atmospheric BrC at night following a large-scale bonfire event. Bluvshtein et al. (Citation2017) reported an increase in k values of the (biomass-burning dominated) atmospheric BrC in the mid-visible range in the night hours following the bonfire event, suggesting enhanced BrC absorption due to nighttime aging. Lin et al. (Citation2017) reported that unlike fresh biomass-burning BrC chromophores which were dominated by aromatic compounds with no nitro groups (Lin et al. Citation2016), the nighttime aged BrC chromophores were dominated by nitroaromatic species that were formed from NO3 oxidation of aromatic precursors. The evidence from these two studies points to the importance of the enhancement of BrC absorption due to nighttime reactions with NO3. Our controlled combustion and NO3 oxidation experiments provide support for these atmospheric observations and motivate the need for further systematic atmospheric measurements that target the nighttime aging of BrC.
4. Conclusions
We performed toluene-combustion experiments controlled at different combustion conditions to produce BrC with varying light-absorption properties, which we categorized as light, medium, and dark BrC. By exposing the BrC to NO3 in an OFR, we investigated the evolution of the BrC chemical composition and light-absorption properties due to NO3-induced oxidation. The BrC evolved chemically due to both heterogeneous oxidation of the particulate PAHs as well as SOA formation driven by gas-phase oxidation and subsequent condensation of PAH vapors. We performed calculations to decouple the effects of heterogeneous oxidation and SOA formation on the evolution of the light-absorption properties of the BrC and showed that heterogeneous oxidation added chromophoric functional groups that significantly increased light absorption by all 3 BrC types. On the other hand, the oxidized PAHs that contributed to SOA were likely less absorbing than the particulate PAHs, thereby reducing the overall light absorption of the oxidized BrC. Unlike with the light and medium BrC experiments, SOA formation was prominent in the dark BrC experiments, which was manifested as a slight decrease in overall absorption upon oxidation with NO3. However, this effect may have been specific to experimental conditions that suppressed the relative importance of heterogenous oxidation processes because of the short OFR residence time. NO3-induced heterogeneous reactions are expected to be more important than gas-phase reactions at atmospheric conditions. Therefore, nighttime oxidation with NO3 is expected to enhance the light absorption of some atmospheric BrC species.
Supplemental Material
Download MS Word (372.2 KB)Acknowledgments
The authors gratefully thank Manjula Canagaratna, Philip Croteau, Douglas Worsnop, John Jayne, Joseph Roscioli, and David Nelson (ARI) for helpful discussions.
Additional information
Funding
References
- Adachi, K., A. J. Sedlacek, III, L. Kleinman, D. Chand, M. John, P. R. Buseck. 2018. Volume changes upon heating of aerosol particles from biomass burning using transmission electron microscopy. Aerosol Sci. Technol. 52 (1):46–56. doi:10.1080/02786826.2017.1373181.
- Adler, G., N. L. Wagner, K. D. Lamb, K. M. Manfred, J. P. Schwarz, A. Franchin, A. M. Middlebrook, R. A. Washenfelder, C. C. Womack, R. J. Yokelson, et al. 2019. Evidence in biomass burning smoke for a light- absorbing aerosol with properties intermediate between brown and black carbon. Aerosol Sci. Technol. 53 (9):976–89. doi:10.1080/02786826.2019.1617832.
- Aiken, A. C., P. F. Decarlo, and J. L. Jimenez. 2007. Elemental analysis of organic species with electron ionization high-resolution mass spectrometry. Anal. Chem. 79 (21):8350–8. doi:10.1021/ac071150w.
- Atkinson, R. 1991. Kinetics and mechanisms of the gas-phase reactions of the NO3 radical with organic compounds. J. Phys. Chem. Ref. Data 20 (3):459–506. doi:10.1063/1.555887.
- Atkinson, R., J. Arey, B. Zielinska, and S. M. Aschmann. 1990. Kinetics and nitro-products of the gas-phase OH and NO3 radical-initiated reactions of naphthalene-D8 fluoranthene-D10, and pyrene. Int. J. Chem. Kinet. 22 (9):999–1014. doi:10.1002/kin.550220910.
- Bandowe, B. A. M., and H. Meusel. 2017. Nitrated polycyclic aromatic hydrocarbons (Nitro-PAHs) in the environment—A review. Sci. Total Environ. 581–582:237–57. doi:10.1016/j.scitotenv.2016.12.115.
- Bluvshtein, N., P. Lin, J. M. Flores, L. Segev, Y. Mazar, E. Tas, G. Snider, C. Weagle, S. S. Brown, A. Laskin, et al. 2017. Broadband optical properties of biomass-burning aerosol and identification of brown carbon chromophores. J. Geophys. Res. Atmos. 122 (10):5441–56. doi:10.1002/2016JD026230.
- Brown, H., X. Liu, Y. Feng, Y. Jiang, M. Wu, Z. Lu, C. Wu, S. Murphy, and R. Pokhrel. 2018. Radiative effect and climate impacts of brown carbon with the community atmosphere model (CAM5). Atmos. Chem. Phys. 18 (24):17745–68. doi:10.5194/acp-18-17745-2018.
- Browne, E. C., X. Zhang, J. P. Franklin, K. J. Ridley, T. W. Kirchstetter, K. R. Wilson, C. D. Cappa, and J. H. Kroll. 2019. Effect of heterogeneous oxidative aging on light absorption by biomass burning organic aerosol. Aerosol Sci. Technol. 53 (6):663–74. doi:10.1080/02786826.2019.1599321.
- Bruns, E. A., V. Perraud, A. Zelenyuk, M. J. Ezell, S. N. Johnson, Y. Yu, D. Imre, B. J. Finlayson-Pitts, and M. L. Alexander. 2010. Comparison of FTIR and particle mass spectrometry for the measurement of particulate organic nitrates. Environ. Sci. Technol. 44 (3):1056–61. doi:10.1021/es9029864.
- Canagaratna, M. R., J. L. Jimenez, J. H. Kroll, Q. Chen, S. H. Kessler, P. Massoli, L. Hildebrandt Ruiz, E. Fortner, L. R. Williams, K. R. Wilson, et al, 2015. Elemental ratio measurements of organic compounds using aerosol mass spectrometry: Characterization, improved calibration, and implications. Atmos. Chem. Phys. 15 (1):253–72. doi:10.5194/acp-15-253-2015.
- Cheng, Z., K. Atwi, T. Onyima, and R. Saleh. 2019. Investigating the dependence of light-absorption properties of combustion carbonaceous aerosols on combustion conditions. Aerosol Sci. Technol. 53 (4):419–34. doi:10.1080/02786826.2019.1566593.
- Claeys, M., R. Vermeylen, F. Yasmeen, Y. Gómez-González, X. Chi, W. Maenhaut, T. Mészáros, and I. Salma. 2012. Chemical characterisation of humic-like substances from urban, rural and tropical biomass burning environments using liquid chromatography with UV/Vis photodiode array detection and electrospray ionisation mass spectrometry. Environ. Chem. 9 (3):273–84. doi:10.1071/EN11163.
- Dasari, S., A. Andersson, S. Bikkina, H. Holmstrand, K. Budhavant, S. Satheesh, E. Asmi, J. Kesti, J. Backman, A. Salam, et al. 2019. Photochemical degradation affects the light absorption of water-soluble brown carbon in the South Asian outflow. Sci. Adv. 5 (1):eaau8066. doi:10.1126/sciadv.aau8066.
- DeCarlo, P. F., J. R. Kimmel, A. Trimborn, M. J. Northway, J. T. Jayne, A. C. Aiken, M. Gonin, K. Fuhrer, T. Horvath, K. S. Docherty, et al. 2006. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 78 (24):8281–9. doi:10.1021/ac061249n.
- Desyaterik, Y., Y. Sun, X. Shen, T. Lee, X. Wang, T. Wang, and J. L. Collett, Jr. 2013. Speciation of “brown” carbon in cloud water impacted by agricultural biomass burning in Eastern China. J. Geophys. Res. Atmos. 118 (13):7389–99. doi:10.1002/jgrd.50561.
- Farmer, D. K., A. Matsunaga, K. S. Docherty, J. D. Surratt, J. H. Seinfeld, P. J. Ziemann, and J. L. Jimenez. 2010. Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry. Proc. Natl. Acad. Sci. USA. 107 (15):6670–5. doi:10.1073/pnas.0912340107.
- Feng, Y., V. Ramanathan, and V. R. Kotamarthi. 2013. Brown carbon: A significant atmospheric absorber of solar radiation? Atmos. Chem. Phys. 13 (17):8607–21. doi:10.5194/acp-13-8607-2013.
- Forrister, H., J. Liu, E. Scheuer, J. Dibb, L. Ziemba, K. L. Thornhill, B. Anderson, G. Diskin, A. E. Perring, J. P. Schwarz, et al. 2015. Evolution of brown carbon in wildfire plumes. Geophys. Res. Lett. 42 (11):4623–30. doi:10.1002/2015GL063897.
- Hammer, M. S., R. V. Martin, A. Van Donkelaar, V. Buchard, O. Torres, D. A. Ridley, and R. J. D. Spurr. 2016. Interpreting the ultraviolet aerosol index observed with the OMI satellite instrument to understand absorption by organic aerosols: Implications for atmospheric oxidation and direct radiative effects. Atmos. Chem. Phys. 16 (4):2507–23. doi:10.5194/acp-16-2507-2016.
- Heald, C. L., J. H. Kroll, J. L. Jimenez, K. S. Docherty, P. F. Decarlo, A. C. Aiken, Q. Chen, S. T. Martin, D. K. Farmer, and P. Artaxo. 2010. A simplified description of the evolution of organic aerosol composition in the atmosphere. Geophys. Res. Lett. 37 (8):L08803. doi:10.1029/2010GL042737.
- Hoffer, A., A. Tóth, I. Nyirő-Kósa, M. Pósfai, and A. Gelencsér. 2016. Light absorption properties of laboratory-generated tar ball particles. Atmos. Chem. Phys. 16 (1):239–46. doi:10.5194/acp-16-239-2016.
- Jariyasopit, N., M. Mcintosh, K. Zimmermann, J. Arey, R. Atkinson, P. H. Cheong, R. G. Carter, T. Yu, R. H. Dashwood, and S. L. M. Simonich. 2014. Novel nitro-PAH formation from heterogeneous reactions of PAHs with NO2, NO3/N2O5, and OH radicals: Prediction, laboratory studies, and mutagenicity. Environ. Sci. Technol. 48 (1):412–9. doi:10.1021/es4043808.
- Jethva, H., and O. Torres. 2011. Satellite-based evidence of wavelength-dependent aerosol absorption in biomass burning smoke inferred from ozone monitoring instrument. Atmos. Chem. Phys. 11 (20):10541–51. doi:10.5194/acp-11-10541-2011.
- Jo, D. S., R. J. Park, S. Lee, S. Kim, and X. Zhang. 2016. A global simulation of brown carbon: Implications for photochemistry and direct radiative effect. Atmos. Chem. Phys. 16 (5):3413–32. doi:10.5194/acp-16-3413-2016.
- Karavalakis, G., V. Boutsika, S. Stournas, and E. Bakeas. 2011. Biodiesel emissions profile in modern diesel vehicles. Part 2: Effect of biodiesel origin on carbonyl, PAH, nitro-PAH and oxy-PAH emissions. Sci. Total Environ. 409 (4):738–47. doi:10.1016/j.scitotenv.2010.11.010.
- Keyte, I. J., R. M. Harrison, and G. Lammel. 2013. Chemical reactivity and long-range transport potential of polycyclic aromatic hydrocarbons – A review. Chem. Soc. Rev. 42 (24):9333–91. doi:10.1039/c3cs60147a.
- Knopf, D. A., S. M. Forrester, and J. H. Slade. 2011. Heterogeneous oxidation kinetics of organic biomass burning aerosol. Phys. Chem. Chem. Phys. 13 (47):21050–62. doi:10.1039/c1cp22478f.
- Krechmer, J., F. Lopez-Hilfiker, A. Koss, M. Hutterli, C. Stoermer, B. Deming, J. Kimmel, C. Warneke, R. Holzinger, J. Jayne, et al. 2018. Evaluation of a new reagent-ion source and focusing ion-molecule reactor for use in proton-transfer-reaction mass spectrometry. Anal. Chem. 90 (20):12011–8. doi:10.1021/acs.analchem.8b02641.
- Kwamena, N. A., and J. P. D. Abbatt. 2008. Heterogeneous nitration reactions of polycyclic aromatic hydrocarbons and n-hexane soot by exposure to NO3/NO2/N2O5. Atmos. Environ. 42 (35):8309–14. doi:10.1016/j.atmosenv.2008.07.037.
- Kwon, D., M. J. Sovers, V. H. Grassian, P. D. Kleiber, and M. A. Young. 2018. Optical properties of humic material standards: Solution phase and aerosol measurements. ACS Earth Space Chem. 2 (11):1102–11. doi:10.1021/acsearthspacechem.8b00097.
- Lack, D. A., J. M. Langridge, R. Bahreini, C. D. Cappa, A. M. Middlebrook, and J. P. Schwarz. 2012. Brown carbon and internal mixing in biomass burning particles. Proc. Natl. Acad. Sci. USA. 109 (37):14802–7. doi:10.1073/pnas.1206575109.
- Lambe, A. T., A. T. Ahern, L. R. Williams, J. G. Slowik, J. P. S. Wong, J. P. D. Abbatt, W. H. Brune, N. L. Ng, J. P. Wright, D. R. Croasdale, et al. 2011. Characterization of aerosol photooxidation flow reactors: Heterogeneous oxidation, secondary organic aerosol formation and cloud condensation nuclei activity measurements. Atmos. Meas. Tech. 4 (3):445–61. doi:10.5194/amt-4-445-2011.
- Lambe, A. T., E. C. Wood, J. E. Krechmer, F. Majluf, L. R. Williams, L. Croteau, M. Cirtog, A. Féron, J.-E. Petit, A. Albinet, et al. 2020. Nitrate radical generation via continuous generation of dinitrogen pentoxide in a laminar flow reactor coupled to an oxidation flow reactor. Atmos. Meas. Tech. Discuss. doi:10.5194/amt-2019-470.
- Laskin, A., J. Laskin, and S. A. Nizkorodov. 2015. Chemistry of atmospheric brown carbon. Chem. Rev. 115 (10):4335–82. doi:10.1021/cr5006167.
- Lee, H. J., P. K. Aiona, A. Laskin, J. Laskin, and S. A. Nizkorodov. 2014. Effect of solar radiation on the optical properties and molecular composition of laboratory proxies of atmospheric brown carbon. Environ. Sci. Technol. 48 (17):10217–26. doi:10.1021/es502515r.
- Li, C., Q. He, A. P. S. Hettiyadura, U. KäFer, G. Shmul, D. Meidan, R. Zimmermann, S. S. Brown, C. George, A. Laskin, et al. 2019. Formation of secondary brown carbon in biomass burning aerosol proxies through NO3 radical reactions. Environ. Sci. Technol. 54 (3):1395–1405. doi:10.1021/acs.est.9b05641.
- Li, C., Q. He, J. Schade, J. Passig, R. Zimmermann, D. Meidan, A. Laskin, and Y. Rudich. 2019. Dynamic changes in optical and chemical properties of tar ball aerosols by atmospheric photochemical aging. Atmos. Chem. Phys. 19 (1):139–63. doi:10.5194/acp-19-139-2019.
- Lin, P., P. K. Aiona, Y. Li, M. Shiraiwa, J. Laskin, S. A. Nizkorodov, and A. Laskin. 2016. Molecular Characterization of brown carbon in biomass burning aerosol particles. Environ. Sci. Technol. 50 (21):11815–24. doi:10.1021/acs.est.6b03024.
- Lin, P., N. Bluvshtein, Y. Rudich, S. A. Nizkorodov, J. Laskin, and A. Laskin. 2017. Molecular chemistry of atmospheric brown carbon inferred from a nationwide biomass burning event. Environ. Sci. Technol. 51 (20):11561–70. doi:10.1021/acs.est.7b02276.
- Liu, C., P. Zhang, B. Yang, Y. Wang, and J. Shu. 2012. Kinetic studies of heterogeneous reactions of polycyclic aromatic hydrocarbon aerosols with NO3 radicals. Environ. Sci. Technol. 46 (14):7575–80. doi:10.1021/es301403d.
- Liu, D., T. Lin, J. H. Syed, Z. Cheng, Y. Xu, K. Li, G. Zhang, and J. Li. 2017. Concentration, source identification, and exposure risk assessment of PM2.5-bound parent PAHs and nitro-PAHs in atmosphere from typical Chinese cities. Sci. Rep. 7 (1):10398. doi:10.1038/s41598-017-10623-4.
- Lu, J. W., J. M. Flores, A. Lavi, A. Abo-Riziq, and Y. Rudich. 2011. Changes in the optical properties of benzo[a]pyrene-coated aerosols upon heterogeneous reactions with NO2 and NO3. Phys. Chem. Chem. Phys. 13 (14):6484–92. doi:10.1039/c0cp02114h.
- Mak, J., S. Gross, and A. K. Bertram. 2007. Uptake of NO3 on soot and pyrene surfaces. Geophys. Res. Lett 34 (10):L10804. doi:10.1029/2007GL029756.
- Malloy, Q. G. J., S. Nakao, L. Qi, R. Austin, C. Stothers, H. Hagino, and D. R. Cocker. 2009. Real-time aerosol density determination utilizing a modified scanning mobility particle sizer aerosol particle mass analyzer system. Aerosol Sci. Technol. 43 (7):673–8. doi:10.1080/02786820902832960.
- Michelsen, H. A. 2017. Probing soot formation, chemical and physical evolution, and oxidation: A review of in situ diagnostic techniques and needs. Proc. Combust. Inst. 36 (1):717–35. doi:10.1016/j.proci.2016.08.027.
- Moise, T., J. M. Flores, and Y. Rudich. 2015. Optical properties of secondary organic aerosols and their changes by chemical processes. Chem. Rev. 115:4400–39. doi:10.1021/cr5005259.
- Mukai, H., and Y. Ambe. 1986. Characterization of a humic acid-like brown substance in airborne particulate matter and tentative identification of its origin. Atmos. Environ. 20 (5):813–9. doi:10.1016/0004-6981(86)90265-9.
- Nakayama, T., K. Sato, Y. Matsumi, T. Imamura, A. Yamazaki, and A. Uchiyama. 2013. Wavelength and NOx dependent complex refractive index of SOAs generated from the photooxidation of toluene. Atmos. Chem. Phys. 13 (2):531–45. doi:10.5194/acp-13-531-2013.
- Olson, M. R., M. V. Garcia, M. A. Robinson, P. Van Rooy, M. A. Dietenberger, M. Bergin, and J. J. Schauer. 2015. Investigation of black and brown carbon multiple-wavelength- dependent light absorption from biomass and fossil fuel combustion source emissions. J. Geophys. Res. Atmos. 120 (13):6682–97. doi:10.1002/2014JD022970.
- Onasch, T. B., E. C. Fortner, A. M. Trimborn, A. T. Lambe, A. J. Tiwari, L. C. Marr, J. C. Corbin, A. A. Mensah, L. R. Williams, P. Davidovits, et al. 2015. Investigations of SP-AMS carbon ion distributions as a function of refractory black carbon particle type investigations of SP-AMS carbon ion distributions as a function of refractory black carbon particle type. Aerosol Sci. Technol. 49 (6):409–22. doi:10.1080/02786826.2015.1039959.
- Onasch, T. B., A. Trimborn, E. C. Fortner, J. T. Jayne, G. L. Kok, L. R. Williams, P. Davidovits, and D. R. Worsnop. 2012. Soot particle aerosol mass spectrometer: Development, validation, and initial application. Aerosol Sci. Technol. 46 (7):804–17. doi:10.1080/02786826.2012.663948.
- Phillips, S. M., and G. D. Smith. 2014. Light absorption by charge transfer complexes in brown carbon aerosols. Environ. Sci. Technol. Lett. 1 (10):382–6. doi:10.1021/ez500263j.
- Posfai, M., A. Gelencser, R. Simonics, K. Arato, J. Li, P. V. Hobbs, and P. R. Buseck. 2004. Atmospheric tar balls: Particles from biomass and biofuel burning. J. Geophys. Res 109:D06213. doi:10.1029/2003JD004169.
- Saleh, R., Z. Cheng, and K. Atwi. 2018. The brown–black continuum of light-absorbing combustion aerosols. Environ. Sci. Technol. Lett. 5 (8):508–13. doi:10.1021/acs.estlett.8b00305.
- Saleh, R., M. Marks, J. Heo, P. J. Adams, N. M. Donahue, and A. L. Robinson. 2015. Contribution of brown carbon and lensing to the direct radiative effect of carbonaceous aerosols from biomass and biofuel burning emissions. J. Geophys. Res. Atmos. 120 (10):10285–96. doi:10.1002/2015JD023697.
- Saleh, R., E. S. Robinson, D. S. Tkacik, A. T. Ahern, S. Liu, A. C. Aiken, R. C. Sullivan, A. A. Presto, M. K. Dubey, R. J. Yokelson, et al. 2014. Brownness of organics in aerosols from biomass burning linked to their black carbon content. Nat. Geosci. 7 (9):647–50. doi:10.1038/ngeo2220.
- Sarkar, C., C. Venkataraman, S. Yadav, H. C. Phuleria, and A. Chatterjee. 2019. Origin and properties of soluble brown carbon in freshly emitted and aged ambient aerosols over an urban site in India. Environ. Pollut. 254:113077. doi:10.1016/j.envpol.2019.113077.
- Sasaki, J., S. M. Aschmann, E. S. C. Kwok, R. Atkinson, and J. Arey. 1997. Products of the gas-phase OH and NO3 radical-initiated reactions of naphthalene. Environ. Sci. Technol. 31 (11):3173–9. doi:10.1021/es9701523.
- Satish, R., P. Shamjad, N. Thamban, S. Tripathi, and N. Rastogi. 2017. Temporal characteristics of brown carbon over the central Indo- Gangetic Plain. Environ. Sci. Technol. 51 (12):6765–72. doi:10.1021/acs.est.7b00734.
- Sedlacek, A. J., III, P. R. Buseck, K. Adachi, T. B. Onasch, S. R. Springston, and L. Kleinman. 2018. Formation and evolution of tar balls from northwestern US wildfires. Atmos. Chem. Phys. 18 (15):11289–301. doi:10.5194/acp-18-11289-2018.
- Shen, G., S. Tao, S. Wei, Y. Zhang, R. Wang, B. Wang, W. Li, H. Shen, Y. Huang, Y. Chen, et al. 2012. Emissions of parent, nitro, and oxygenated polycyclic aromatic hydrocarbons from residential wood combustion in rural China. Environ. Sci. Technol. 46 (15):8123–30. doi:10.1021/es301146v.
- Sumlin, B. J., A. Pandey, M. J. Walker, R. S. Pattison, B. J. Williams, and R. K. Chakrabarty. 2017. Atmospheric Photooxidation diminishes light absorption by primary brown carbon aerosol from biomass burning. Environ. Sci. Technol. Lett. 4 (12):540–5. doi:10.1021/acs.estlett.7b00393.
- Tóth, A., A. Hoffer, I. Nyirő-Kósa, M. Pósfai, and A. Gelencsér. 2014. Atmospheric tar balls: Aged primary droplets from biomass burning? Atmos. Chem. Phys. 14 (13):6669–75. doi:10.5194/acp-14-6669-2014.
- Utry, N., T. Ajtai, á Filep, M. Dániel Pintér, A. Hoffer, Z. Bozoki, and G. Szabó. 2013. Mass specific optical absorption coefficient of HULIS aerosol measured by a four-wavelength photoacoustic spectrometer at NIR, VIS and UV wavelengths. Atmos. Environ. 69:321–4. doi:10.1016/j.atmosenv.2013.01.003.
- Wang, L., Z. Li, Q. Tian, Y. Ma, F. Zhang, Y. Zhang, D. Li, K. Li, and L. Li. 2013. Estimate of aerosol absorbing components of black carbon, brown carbon, and dust from ground-based remote sensing data of sun-sky radiometers. J. Geophys. Res. Atmos. 118 (12):6534–43. doi:10.1002/jgrd.50356.
- Wang, X., C. L. Heald, D. A. Ridley, J. P. Schwarz, J. R. Spackman, A. E. Perring, H. Coe, D. Liu, and A. D. Clarke. 2014. Exploiting simultaneous observational constraints on mass and absorption to estimate the global direct radiative forcing of black carbon and brown carbon. Atmos. Chem. Phys. 14 (20):10989–10. doi:10.5194/acp-14-10989-2014.
- Wang, X., C. L. Heald, A. J. Sedlacek, S. S. De Sá, S. T. Martin, M. L. Alexander, T. B. Watson, A. C. Aiken, S. R. Springston, and P. Artaxo. 2016. Deriving brown carbon from multiwavelength absorption measurements: Method and application to AERONET and aethalometer observations. Atmos. Chem. Phys. 16 (19):12733–52. doi:10.5194/acp-16-12733-2016.
- Wang, Y., M. Hu, P. Lin, T. Tan, M. Li, N. Xu, J. Zheng, Z. Du, Y. Qin, Y. Wu, et al. 2019. Enhancement in particulate organic nitrogen and light absorption of humic-like substances over tibetan plateau due to long-range transported biomass burning emissions. Environ. Sci. Technol. 53 (24):14222–32. doi:10.1021/acs.est.9b06152.
- Wang, Y., P. L. Ma, J. Peng, R. Zhang, J. H. Jiang, R. C. Easter, and Y. L. Yung. 2018. Constraining aging processes of black carbon in the community atmosphere model using environmental chamber measurements. J. Adv. Model. Earth Syst. 10 (10):2514–26. doi:10.1029/2018MS001387.
- Wong, J. P. S., A. Nenes, and R. J. Weber. 2017. Changes in light absorptivity of molecular weight separated brown carbon due to photolytic aging. Environ. Sci. Technol. 51 (15):8414–21. doi:10.1021/acs.est.7b01739.
- Wong, J. P. S., M. Tsagkaraki, I. Tsiodra, N. Mihalopoulos, K. Violaki, M. Kanakidou, J. Sciare, A. Nenes, and R. J. Weber. 2019. Atmospheric evolution of molecular-weight-separated brown carbon from biomass burning. Atmos. Chem. Phys. 19 (11):7319–34. doi:10.5194/acp-19-7319-2019.
- You, R., J. G. Radney, M. R. Zachariah, and C. D. Zangmeister. 2016. Measured wavelength-dependent absorption enhancement of internally mixed black carbon with absorbing and nonabsorbing materials. Environ. Sci. Technol. 50 (15):7982–90. doi:10.1021/acs.est.6b01473.
- Yu, Z., G. Magoon, J. Assif, W. Brown, and R. Miake-Lye. 2019. A single-pass RGB differential photoacoustic spectrometer (RGB-DPAS) for aerosol absorption measurement at 473, 532, and 671 Nm. Aerosol Sci. Technol. 53 (1):94–105. doi:10.1080/02786826.2018.1551611.
- Zhang, Y., H. Forrister, J. Liu, J. Dibb, B. Anderson, J. P. Schwarz, A. E. Perring, J. L. Jimenez, P. Campuzano-Jost, Y. Wang, et al, 2017. Top-of-atmosphere radiative forcing affected by brown carbon in the upper troposphere. Nature Geosci. 10 (7):486–9. doi:10.1038/ngeo2960.
- Zhang, Y., J. Shu, C. Liu, Y. Zhang, B. Yang, and J. Gan. 2013. Heterogeneous reaction of particle-associated triphenylene with NO3 radicals. Atmos. Environ. 68 (3):114–9. doi:10.1016/j.atmosenv.2012.11.052.
- Zhao, R., A. K. Y. Lee, L. Huang, X. Li, F. Yang, and J. P. D. Abbatt. 2015. Photochemical processing of aqueous atmospheric brown carbon. Atmos. Chem. Phys. 15 (11):6087–100. doi:10.5194/acp-15-6087-2015.
- Zhong, M., and M. Jang. 2014. Dynamic light absorption of biomass-burning organic carbon photochemically aged under natural sunlight. Atmos. Chem. Phys. 14 (3):1517–25. doi:10.5194/acp-14-1517-2014.
- Zielinska, B., J. Arey, R. Atkinson, and P. A. McElroy. 1989. Formation of methylnitronaphthalenes from the gas-phase reactions of 1- and 2-methylnaphthalene with OH radicals and N205 and their occurrence in ambient air. Environ. Sci. Technol. 23 (6):723–9. doi:10.1021/es00064a011.
- Zimmermann, K., N. Jariyasopit, S. L. M. Simonich, S. Tao, R. Atkinson, and J. Arey. 2013. Formation of nitro-PAHs from the heterogeneous reaction of ambient particle-bound PAHs with N2O5/NO3/NO2. Environ. Sci. Technol. 47 (3):8434–−42. doi:10.1021/es401789x.