Abstract
All cases of high-grade osteosarcoma (OS) (n = 196) and Ewing's sarcoma of bone (ES) (n = 56) treated at the Norwegian Radium Hospital in the period 1980–1999 were analyzed retrospectively. They were allocated to consecutive ten-year periods by their time of diagnosis. Patient and tumour characteristics have been relatively stable. Eighty percent of all patients received surgical treatment and the amputation rate decreased from 64% to 23%. The percentage of patients receiving chemotherapy has remained around 80%. The use of radiotherapy in primary treatment decreased gradually from 33% to 18%. Sarcoma specific survival (SSS) at five years for all patients increased significantly from 39% to 53%. Similar trends for improvement were seen for both OS and ES. In multivariate analysis, independent prognostic factors for improved SSS were non-metastatic disease at diagnosis, age under 40, extremity tumours, small tumours and treatment from 1995 onwards. No major new treatment options have emerged over these 20 years. The improved outcome appears partly to be due to refinements in the use of existing modalities and improved quality and integration of multidisciplinary approaches. Improved formalized organisation of the sarcoma group and annual audited reports of its patient and research activity may also have contributed to improved focus and performance.
Bone sarcomas constitute a rare and heterogeneous group of tumours representing approximately 0.2% of all cancers. The incidence is relatively stable and approximately 40 new bone sarcomas occur annually in Norway. Of these 12–15 are osteosarcomas and 5–8 belong to the Ewing family of tumours.
Successful treatment requires high levels of expertise and multidisciplinary management. For patients with highly malignant tumours, the prognosis following surgery alone is poor with survival in the 10–20% range. After the introduction of neoadjuvant chemotherapy in the late 70's and early 80's survival improved dramatically, and during the 80's improvement in surgical techniques at major centres has led to a substantial decrease in amputation rate.
In Norway the diagnosis and treatment of sarcomas is centralized to four regional tumour centres. The Norwegian Radium Hospital (NRH) has the formal role of a National Centre of Reference since 1991, and has traditionally treated around 70% of the Norwegian bone sarcoma patients.
This study is a retrospective analysis of all high grade osteosarcomas and Ewing's sarcoma treated at the NRH from 1980 to 1999, with focus on time trends in patient volume, referral patterns, patient characteristics, treatment and outcome, as well as on development of the NRH sarcoma program.
Patients and methods
The patient database
The NRH sarcoma database contains all sarcoma patients referred to the NRH from January 1980 onwards, and was established in 1994. For the period 1980–85 patients were entered into the database following retrospective review of medical records after identification from NRH's diagnostic registry. For the period 1986–1994 electronic records for NHR patients entered in the Scandinavian Sarcoma Group (SSG) Central Register Citation[1] were imported into the NRH database with subsequent review of the data set, and missing patients were supplemented from the diagnostic registry. Patients treated from 1995 onwards have been entered into the database consecutively, with supplementation from the diagnostic registry at the end of each year. Although secretarial staff has aided in data registration, all data regarding tumour classification, staging, treatment and outcome have been verified by senior clinicians belonging to the NRH sarcoma group.
In the analyses of trends over time the patients were allocated to consecutive 5- or 10-year periods by the time of diagnosis. In some analyses OS and ES were considered together in order to have sufficient patient numbers.
Patient characteristics
From January 1980 until December 1999, a total of 252 patients were treated. Of these patients, 172 had osteosarcoma, 24 had malignant fibrous histiocytoma (MFH) (together constituting the OS group = 196 patients); 51 had Ewing's sarcoma and 5 had primitive neuroectodermal tumour (PNET) (together constituting the ES group = 56 patients). One hundred and sixty three (65%) had extremity tumours and 89 (35%) had axial or pelvic tumours (). Non-extremity tumours were more common for ES (59%) than for OS (29%). The median age at diagnosis was 17 years (range 2–39) for ES and 23 years (6–87) for OS patients, and the overall male/female ratio was 1.43 and 1.58, respectively. Twenty-one percent of ES patients and 25% of OS patients had detectable metastases at the time of diagnosis, and these figures were stable over time. Apart from an increase in the fraction of non-extremity tumours (from 30% to 43%) and an increase in the fraction of female patients, the patient characteristics were relatively stable over the entire study period (1980–99). There was also a slight decrease in the number of OS patients in the last 10-year period ().
Table I. Patient characteristics.
Following primary treatment most patients have been followed up at NRH, and median follow-up time for surviving patients was 218 months (range 21–281) and 96 months (range 59–171) months for the two 10-year periods, respectively.
Multidisciplinary evaluation of individual cases.
For diagnosis and pathology review, staging and choice of treatment, each patient was evaluated by NHR's multidisciplinary sarcoma group, which includes dedicated specialists in orthopaedic, thoracic, abdominal and head and neck surgery, medical oncology, pathology, radiotherapy, diagnostic radiology and nuclear medicine. Specialists in cytogenetics and molecular genetics were included from 1995. To enable a close interaction between the surgeons and the other group members the orthopaedic tumour surgery (and surgeons) was permanently transferred to NRH from Sophies Minde Orthopaedic Hospital in 1996. The group has had weekly tumour boards with display of all relevant histological and radiological material, and all decisions regarding diagnosis, staging and treatment have been taken by the group as a whole. This board also evaluates all patients with soft tissue sarcomas and all benign sarcoma-like conditions in bone and soft tissues. In the tumour board evaluation, a curative treatment attempt for OS and ES was defined as multimodality strategy including combination chemotherapy with surgery and/or radiotherapy with the purpose of elimination of all identifiable tumour with clear macroscopic margins.
Although this basic structure of the group has remained unchanged during the study period, distinctive gradual developments have been made through at least duplication of specialists in all functions to secure continuous expertise, formalization of the responsibility for quality within each speciality, a continuously updated patient database (from 1994) and yearly audited reports of all patient and research activities (from 1995). The group's clinical and laboratory research was substantially increased from 1990 onwards, with emphasis on collaborative efforts with the Scandinavian and Italian Sarcoma Groups Citation[2]. Clinical, laboratory and translational research has gradually become a strong integrated core activity within the sarcoma program.
Histology
As practiced by The Scandinavian Sarcoma Group (SSG), a four-grade malignancy scale was used where grades I and II are considered low-grade and grades III and IV are considered high grade tumours Citation[3], Citation[4]. Only high-grade tumours are included in this paper. Members of the SSG Pathology Review Board (AES and/or BB) have evaluated all tumours histologically. Suspected Ewing's sarcomas were routinely evaluated by immunohistochemistry (a broad panel of antibodies including CD 99 (MIC 2)), and from 1995 also by cytogenetics and/or PCR. All tumours from patients included in prospective clinical SSG studies have had pathology review by the central SSG pathology board. In the current study only bone tumours of high-grade malignancy were included.
Chemotherapy intensity
Chemotherapy for OS and ES was based on the successive prospective study protocols run by the Scandinavian Sarcoma Group (SSG) and its collaborators. For patients not eligible for these studies, the treatment was modified for age, organ function and toxicity with basis in these protocols. Before 1990 chemotherapy for OS was based on the T-10 protocol using single agent high-dose methotrexate (HDMTX) in the preoperative phase and combinations of cyclophosphamide, doxorubicin, cisplatin and dactinomycin in the postoperative phase depending on histological tumour response (SSG II study) Citation[5]. From 1990 the MTX dose was increased, cisplatin and doxorubicin were added in the preoperative treatment, and ifosfamide and etoposide were introduced for poor histological responders (SSG VIII study) Citation[6], Citation[7]. From 1997 the dose levels of several of these agents were increased further, and high-dose ifosfamide was added from the start of treatment (ISG/SSG I study) Citation[6], Citation[8], Citation[9]. In the present analysis, the T-10 regimen and its modifications were classified as of conventional intensity, whereas the SSG VIII, ISG/SSG I and their modifications were classified as regimens of high intensity.
Prior to 1990 patients with ES received conventional multiagent combination chemotherapy of moderate intensity (cyclofosfamide, doxorubicin, methotreaxate, bleomycin, dactinomycin and vincristine), and radiotherapy was given separately following the completion of chemotherapy (SSG IV) Citation[10]. The subsequent SSG IX study added ifosfamide from the start of treatment, the local treatment was given earlier and the radiotherapy schedule was modified and integrated during chemotherapy (see below) Citation[11]. In the present analysis, the SSG IV and similar regimens for Ewing's sarcoma were classified as of conventional intensity, whereas the SSG IX and similar regimens were classified as of high intensity.
Statistical analysis
The statistical package SPSS 12.0.1 for Windows was used for all analyses. For the analysis of possible changes over the time periods, the Chi Square test for linear trends was used. Survival was calculated by the Kaplan Meier method and survival comparisons were performed by the log rank test.
Sarcoma specific survival was defined as the time from diagnosis to death by sarcoma or by treatment complications. In the analysis of metastasis-free survival only patients with non-metastatic disease at diagnosis were included. Cox's proportional hazards model was used to identify factors of individual importance for sarcoma specific survival
Results
Referral pattern and surgical treatment
Eighty percent of all patients (and 85% of patients with localized disease) were treated by surgery, most of these in combination with chemotherapy (69%), radiotherapy (3%) or both (14%). Fourteen percent were treated with surgery alone. Thirteen percent of all patients had undergone some form of open surgery before referral to NRH, this proportion decreased from 16% in the first 10-year period to 9% in the second 10-year period. Approximately 90% of extremity tumours were treated with surgery throughout the study periods, and the rare avoidance of surgery in this group was due to extensive metastatic disease, poor performance status or old age. As regards axial tumours, the percentage undergoing surgery increased from 51% to 76% from the first to the last period, the increase being particularly marked for ES (from 47% to 89%). In parallel the use of radiotherapy for ES decreased from 72% to 32% ().
Improved limb-sparing surgical techniques led to a substantial decrease in amputation rate during the last two decades. Whereas 64% of tumour-bearing extremities (69% of patients with localized disease) were amputated in the first time period, only 23% (25% of patients with localized disease) were amputated in the last ().
Table II. Treatment of non-metastatic osteosarcoma/MFH (n = 146) and Ewing family of tumours (n = 44).
Chemotherapy
Multidisciplinary treatment is mandatory for success in the treatment of osteo- and Ewing's sarcoma. Approximately 80% of all patients received chemotherapy throughout the study period (76% of all OS and 96% of all ES). Chemotherapy intensity increased over time, and from 1990 onwards 84% of all patients received chemotherapy defined as of high intensity, as compared to 7% before 1990. The fraction of OS patients that received protocol-based treatment was 84% and 76% in the subsequent periods, for ES the corresponding figures were 68% and 89%. However, in the two subsequent time periods only 28% and 30% of OS patients, and 57% and 71% of ES patients, were formally included in prospective clinical studies. No eligible patients failed to be included, the reasons for non-inclusion being overt metastases at diagnosis, age > 40 years, axial OS tumours or a time of diagnosis in between the time of activation of formal studies. A higher fraction of non-extremity tumours combined with higher age was also the reason for fewer OS patients being treated on protocol in period 2. In the ES studies, all tumour sites were eligible, and no patients were too old for protocol treatment.
Radiotherapy
The proportion of patients treated with radiotherapy as a part of their initial management has decreased for both tumour types, and the decrease was particularly marked for ES (). For osteosarcoma and MFH the decrease was related to a realization of a limited sensitivity to radiation, whereas for ES the decrease was due to an increase in the use of surgery for axial and pelvic tumours coupled with a documentation of surgical superiority over radiotherapy in achieving local control Citation[12]. For ES patients who received radiotherapy the schedule was intensified in 1990 (protocol SSG IX), with hyperfractionated accelerated radiotherapy being closely integrated with chemotherapy Citation[11].
Treatment of metastatic disease
In total 157 patients developed metastatic disease. Of 62 patients with metastases at diagnosis 81% received combination chemotherapy, whereas re-treatment with chemotherapy was administered to 62% of the 95 patients who suffered metastatic relapse. The fraction of metastatic patients treated with chemotherapy remained stable throughout the study periods. The use of metastasectomy (mainly the resection of lung metastases in OS) increased from 37% to 47% in the last 10-year period, resulting in an increase in patients achieving clinical CR at the first metastatic event from 34% to 50%. This increase was most marked from 1995 onwards, where 62% underwent metastasectomy and 69% achieved a clinical CR. This development was not associated with a decreasing number of detectable metastases (median 5), and from 1995 the increased surgical aggressiveness is illustrated by an increase in the median number of resected metastases from two to six in the patients who achieved a CR.
Sarcoma specific survival
shows a gradual time-dependent increase in sarcoma-specific survival (SSS) for the entire patient population, resulting in a significant improvement from 39% to 53% at five years comparing the two subsequent 10-year periods (p = 0.03). Although the improvement over time was most marked for patients with non-metastatic disease, male gender, Ewing histology and patients who were not included in prospective clinical studies, there was a varying but consistent trend for improved outcome with time for nearly all patient categories (). For localized osteosarcoma 5-year SSS increased from 47% to 59% (p = 0.31), and for localized ES from 36% to 68% (p = 0.03, ).
Figure 1. Sarcoma specific survival for all patients by period of diagnosis.
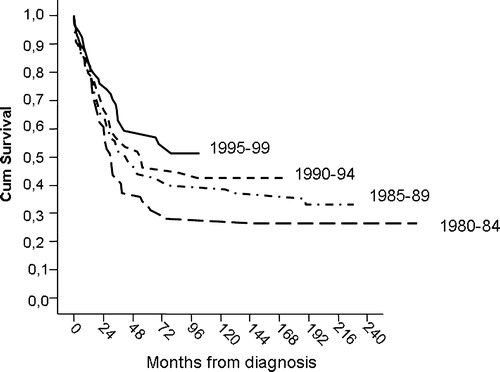
Figure 2. Sarcoma specific survival for non-metastatic osteosarcoma and Ekring's sarcoma by period of diagnosis.
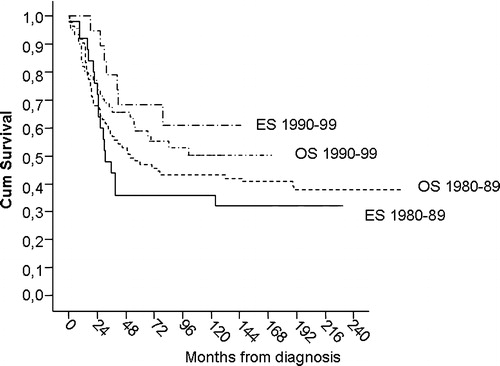
Table III. Development over time in five-year sarcoma-specific survival in different patient and tumour categories.
For patients with synchronous or metachronous metastatic disease the increasing use of metastasectomy did not increase post-metastatic survival for osteosarcoma patients (median 14 months in both time periods, 83 and 41 patients, respectively). However, median survival from the first metastatic event in ES increased dramatically from 8 to 50 months (p = 0.008, 18 and 15 patients in the successive periods)).
Metastasis-free survival
Analogous to the improvement in SSS there was a gradual increase in metastasis-free survival for all patients with initial localized disease from 37% to 62% over the subsequent 10-year periods (p = 0.001). For OS the increase was from 36% to 60% (p = 0.008) and for ES the increase was from 40% to 68% (p = 0.05).
Local recurrence-free survival
For the patient population as a whole the risk of local recurrence decreased from 21% to 11% at 5 years in the two consecutive 10-year periods (p = 0.09). This trend for improvement was present both for ES (24% vs. 6%) and OS (20% vs. 13%), and was more evident for axial tumours (52% vs. 22%, p = 0.03) than extremity tumours (8% vs. 2%, p = 0.14). The significant improvement for axial tumours was probably a result of more extensive use of surgery for axial tumours in the last period (81% in the last period vs. 62% in the first).
Prognostic factors for sarcoma specific survival
In univariate analyses non-metastatic disease at diagnosis, age ≤ 40 years, extremity tumour site, small tumour size, diagnosis after 1990, inclusion in a prospective study, and high chemotherapy intensity were factors associated with significantly improved sarcoma-specific survival. In multivariate analysis, the factors that retained significant predictive value were non-metastatic disease, age ≤ 40, extremity tumour site, small tumour size and treatment from 1995 onwards ().
Table IV. Favourable prognostic factors for sarcoma specific survival (195 OS and ES patients with complete data).
For patients with localized disease multivariate analysis revealed extremity tumour site, small tumour size and inclusion in a prospective clinical study as significant favourable factors, whereas diagnosis from 1995 onwards and age ≤ 40 were of borderline significance (). In localized osteosarcoma the significant factors in multivariate analysis were extremity site (p < 0.001), small tumour size (p = 0.001) and age ≤ 40 (p = 0.02), whereas in Ewing's sarcoma they were participation in a prospective trial (p = 0.04) and diagnosis after 1990 (p = 0.04) or 1995 (p = 0.005). However in these analyses the OS and ES groups consisted of only 123 and 44 patients, respectively.
Table V. Favourable prognostic factors for patients with localized disease at diagnosis (154 OS and ES patients with complete data).
Discussion
Most published studies on osteosarcoma and Ewing's sarcoma apply patient selection on the basis of age, tumour site, stage or treatment protocol, and are thus not representative of OS and ES in general Citation[13]. In order to address a more general development in the treatment and outcome of high-grade bone sarcomas we therefore present an unselected patient material of OS and ES from a single national reference institution, with analyses of general trends over time across all ages, stages and tumour sites, and with sub analyses where appropriate. Patients with MFH and PNET of bone were also included due to their close similarity to OS and ES, respectively, in terms of biological behaviour, treatment approaches and outcome.
Although patient referral to the NRH sarcoma program and patient characteristics have been relatively stable over the study period, there was a slight decrease over time in the referral of osteosarcomas (particularly limb tumours in younger patients), and there was a trend towards a decreasing dominance of male patients and an increase in the fraction of non-extremity tumours (). Female sex may be associated with an improved outcome and non-extremity tumours with a worse outcome, but collectively we do not consider these factors to be of major explanatory value for the improvement in results.
Our analyses document a significant improvement over time in the rate of limb salvage, local tumour control, metastasis-free survival and sarcoma-specific survival. Although patient numbers necessitated the allocation to one of two 10-year periods, shows that the improvement appears to be of a gradual nature.
The multivariate analysis shows that age ≤ 40, localized disease, extremity tumour site and small tumour size were independent factors associated with a favourable sarcoma-specific survival. These are all well established prognosticators in high-grade bone sarcomas. It should be pointed out that the relatively low number of patients prohibited a detailed analysis of sufficient power in OS and ES separately, and although there was a close similarity in the prognostic factors for OS and ES, a pooled analysis of this kind should be interpreted with caution. The age cut-off of 40 years was chosen due to the SSG trials in OS and ES having this as the maximum age for eligibility, leading to an individualization of treatment for older patients. Despite the importance of age, the formal inclusion in prospective trials was associated with an improved survival, particularly for ES patients. The positive impact of participation in clinical trials is well known, and may be due to the meticulous attention, state-of-the-art care and follow up offered to these patients Citation[14]. Although the intensification of chemotherapy has probably contributed to improved outcome it is interesting to note that for both tumour types the participation in trials per se appears more important (). This finding supports the view that no major chemotherapy breakthrough can explain the entire improvement in survival seen over time, and the fraction of patients treated with chemotherapy has remained virtually unchanged (). The international experience is that although major improvements were seen in both OS and ES after the introduction of effective combination chemotherapy in the late 1970s and early 1980s, subsequent further intensification has led to only limited progress Citation[6], Citation[13], Citation[15]. It should also be noted that there is no evidence in the literature of the natural prognosis of OS and ES having changed over the time period in question.
For patients with localized disease the 5-year sarcoma-specific survival for the last 10-year diagnostic period of 59% for OS and 68% for ES are comparable to survival rates reported in the literature, particularly when taking into account that our data includes all ages and tumour sites. The significant improvement in survival over time for patients with localized ES () is in accordance with the improvement that was generally seen in the SSG IX study, where the majority of our patients were included. Although this was not a randomized study the improvement was thought to be due to intensification of chemotherapy with ifosfamide, earlier local treatment with the increased use of surgery, and hyperfractionated radiotherapy closely integrated with chemotherapy Citation[11].
The outcome in metastatic ES has remained very poor despite various treatment attempts with high-dose chemotherapy and stem cell rescue Citation[16]. We were therefore surprised to find a statistically significant improvement in median survival from the first metastatic event from 8 to 50 months, giving an improvement in projected survival at 3 years from 12% to 53% (p = 0.008). There were no differences between the periods as regards metastatic sites or the number of metastases (pulmonary versus skeletal). Six of seven survivors had metastases at diagnosis rather than metastatic relapse, and although the reasons for this improvement are unclear, it may be related to the use of primary consolidation with chemotherapy intensification and irradiation of all identifiable metastatic sites modified from Burdach et al. Citation[17]. This approach was used only for selected patients, and high-dose ifosfamide (15 g/m2) was more often employed than high-dose regimens requiring stem cell rescue. However, our findings are based on only 30 patients with metastatic ES, and must therefore be interpreted with caution. A separate detailed analysis will be performed, extending the study period until 2004.
In contrast with the findings in metastatic ES, no improvement was seen in metastatic OS. This was despite the treatment of metastatic OS being associated with a more aggressive approach towards pulmonary metastasectomy with removal of a larger number of metastases in the last period. This development was inspired by several reports on the importance of complete surgical removal of all identifiable metastases for subsequent survival, but the lack of improvement indicates that the number of metastases is significantly more important Citation[18].
Which other factors may have contributed to the gradual improvement in local recurrence-free, metastasis-free and sarcoma-specific survival, including a uniform positive trend through nearly all patient categories ()? The multivariate analysis of the entire patient population () shows that the last time period retained independent favourable prognostic impact after correction for other major prognostic variables, although maximum tumour extension should ideally have been exchanged for tumour volume Citation[19], and some factors such as the expression of genes conveying chemotherapy resistance have not been analysed. Chemotherapy intensification fell short of statistical significance as it largely coincided with treatment from 1990 onwards, indicating that additional elements in the last decade were of importance for improved outcome ().
Although this is hard to demonstrate clearly, we believe parts of the observed improvement to be due to a general progress in the accumulated expertise, organisation and performance of the multidisciplinary sarcoma group, and particularly in the quality of care for patients ineligible for formal prospective studies Citation[13], Citation[20] (). This does not imply an overall increase in the use of chemotherapy (), and may rather be a result of more successful patient selection based on accumulated experience.
Although of uneven magnitude, the time trend for improved survival appeared to be constant through most patient subgroups and types of treatment. Other unidentified prognostic factors may have caused bias in the patient distribution, and patient numbers were small. Nevertheless we believe that the progress over time indicates a time-dependent general improvement in overall performance by the multidisciplinary sarcoma team. The sarcoma program has undergone a gradual development, including concentration of all functions to one institution, addition of new relevant specialities (cytogenetics, molecular genetics), at least duplication of specialists in all functions, formalized responsibility within each speciality, increased international cooperation, increased focus on integrated clinical and laboratory research, and a continuously updated patient database forming the basis for research and annual audited reports of all the program activities. The positive development in clinical skills is illustrated by the gradual increase in the use of various limb-sparing surgical techniques for extremity tumours, with an amputation rate of 85%, 46%, 31% and 18% in the subsequent 5-year periods. This development did not jeopardize an excellent local control rate (2% in the last period). There was also an increased in the use of surgery for non-extremity tumours (from 51% to 76%), and this was accompanied by a statistically significant reduction in the projected local recurrence rate from 52% to 22% in the last 10-year period. These improvements have taken place while the patient volume has stayed relatively constant, indicating gradually increasing resources per patient.
Considering the complexity of bone sarcoma treatment our patient volume is a concern, with only 12.6 new patients with OS and ES on average being treated yearly. A higher degree of centralization to fewer centres, at least at key time points in the treatment course, may be a way forward.
An improvement in the results for rare tumours on the basis of new therapy is a very gradual and stepwise process. We believe that centralization dedicated expert centres with an adequate patient volume, and organisational refinement of their activities are vital elements in improving the quality of care.
Related Research Data
References
- Bauer HC, Alvegard TA, Berlin O, et al. The Scandinavian Sarcoma Group Register. Acta Orthop Scand 1999; 70(Suppl 285)41–4
- Alvegard, TA, Rydholm, A. The Scandinavian Sarcoma Group. Twenty years experience. Acta Orthop Scand 1999;70, (Suppl 285).
- Unni KK, Dahlin DC. Grading of bone tumors. Semin Diagn Pathol 1984; 1: 165–17
- Inwards CY, Unni KK. Classification and grading of bone sarcomas. Hematol Oncol Clin North Am 1995; 9: 545–69
- Saeter G, Alvegard TA, Elomaa I, Stenwig AE, Holmstrom T, Solheim OP. Treatment of osteosarcoma of the extremities with the T-10 protocol, with emphasis on the effects of preoperative chemotherapy with single-agent high-dose methotrexate: A Scandinavian Sarcoma Group study. J Clin Oncol 1991; 9(10)1766–75
- Saeter G, Wiebe T, Wiklund T, et al. Chemotherapy in osteosarcoma. The Scandinavian Sarcoma Group experience. Acta Orthop Scand 1999; 70(Suppl 285)74–82
- Smeland S, Muller C, Alvegard TA, et al. Scandinavian Sarcoma Group Osteosarcoma Study SSG VIII: Prognostic factors for outcome and the role of replacement salvage chemotherapy for poor histological responders. Eur J Cancer 2003; 39: 488–94
- Bacci G, Ferrari S, Longhi A, et al. High dose ifosfamide in combination with high dose metothrexate, adriamycin and cisplatin in the neoadjuvant treatment of extremity osteosarcoma:preliminary results of an Italian Sarcoma Group/Scandinavian Sarcoma Group pilot study. J Chemother 2002; 14(2)198–206
- Smeland, S, Bacci, G, Ferrari, S, et al. Neoadjuvant chemotherapy with high-dose ifosfamide added to methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity. A joint study by the Italian (ISG) and Scandinavian sarcoma groups. Proceedings of ASCO 2003;22:817, (abstr).
- Nilbert M, Saeter G, Elomaa I, et al. Ewing's sarcoma treatment in Scandinavia 1984–1990–ten-year results of the Scandinavian Sarcoma Group Protocol SSGIV. Acta Oncol 1998; 37: 375–8
- Elomaa I, Blomqvist CP, Saeter G, et al. Five-year results in Ewing's sarcoma. The Scandinavian Sarcoma Group experience with the SSG IX protocol. Eur J Cancer 2000; 36: 875–80
- Rosito P, Mancini AF, Rondelli R, et al. Italian Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone: A preliminary report of 6 years of experience. Cancer 1999; 86: 421–8
- Bruland OS, Pihl A. On the current management of osteosarcoma. A critical evaluation and a proposal for a modified treatment strategy. Eur J Cancer 1997; 33: 1725–31
- Stiller CA. Survival of patients with cancer. Those included in clinical trials do better. BMJ 1989; 299: 1058–9
- Elomaa I, Blomqvist C, Saeter G, et al. Chemotherapy in Ewing's sarcoma. The Scandinavian Sarcoma Group experience. Acta Orthop Scand 1999; 70(Suppl 285)69–73
- Rodriguez-Galindo C, Spunt SL, Pappo AS. Treatment of Ewing sarcoma family oftumors:current status and outlook for the future. Med Pediatr Oncol 2003; 40: 276–87
- Burdach S, Meyer-Bahlburg A, Laws HJ, et al. High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors: Results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 2003; 21: 3072–8
- Saeter G, Høie J, Stenwig AE, et al. Systemic relapse of patients with osteogenic sarcoma. Prognostic factors for long term survival. Cancer 1995; 75: 1084–93
- Gobel V, Jurgens H, Etspuler G, et al. Prognostic significance of tumor volume in localized Ewing's sarcoma of bone in children and adolescents. J Cancer Res Clin Oncol 1987; 113: 187–91
- Sæter G, Bruland ØS, Follerås G, et al. Extremity and non-extremity high-grade osteosarcoma. The Norwegian Radium Hospital experience during the modern chemotherapy era. Acta Oncol 1996; 35(Suppl 8)129–34