Abstract
Many germline mutations in the DNA mismatch repair genes have been described so far leading to the clinical phenotype of Lynch syndrome (hereditary nonpolyposis colorectal cancer, HNPCC). Most mutations are private mutations. We report on nine novel pathogenic germline mutations that have been found in families meeting either the Amsterdam or the Bethesda criteria. These findings include the mutations MLH1,c.884+4A>G, MLH1,c.1377_1378insA;p.Glu460ArgfsX19, MLH1,c.1415_1416delGA;p.Arg472ThrfsX5, MSH2,c.301G>T;p.Glu101X, MSH2,c.638_639delTG;p.Leu213GlnfsX18, MSH2,c.842C>A;p.Ser281X, MSH2,c.859G>T;p.Gly287X, MSH6,c.2503C>T;p.Gln835X and a large genomic deletion of exons 1–10 of the PMS2 gene. The mutation MLH1,c.884+4A>G detected in two families results in a complete skipping of exon 10 on mRNA level and thus has been considered as pathogenic. In all cases the tumor tissue of the index patient revealed high microsatellite instability (MSI-H) and showed a complete loss of expression of the affected protein in the tumor cells by immunohistochemistry (IHC). The findings underline the importance of a pre-screening of tumor tissue for an efficient definition of conspicuous cases.
Lynch syndrome (hereditary non-polyposis colorectal cancer/HNPCC) is one of the most common cancer susceptibility syndromes with an autosomal dominant inheritance caused by germline mutations in DNA mismatch repair (MMR) genes, predominantly MLH1 and MSH2 Citation[1]. In a minority of cases, mutations have been described in the MMR genes MSH6 and PMS2 Citation[2–4]. Mutation carriers are at risk to develop a broad spectrum of malignancies including tumors of the colorectum, endometrium, stomach, small intestine, hepatobiliary system, ureter, renal pelvis, ovary and skin Citation[1], Citation[5]. Here we report on nine novel pathogenic germline mutations identified in patients suspected of HNPCC. Among these, we demonstrate the functional relevance of an unclassified intronic variant in MLH1. Furthermore, the detection of a PMS2 deletion in a large family extends the still limited knowledge on the tumor spectrum associated with PMS2 defects.
Materials and methods
Patients
The patients included in this study were recruited at the University Hospital Bonn, Germany, within a project of the German HNPCC consortium. Patients were referred to this study from other hospitals, institutes of human genetics, private practice physicians, and private practice human geneticists or by self-referral. Patients were enrolled into the study if they fulfilled at least one of the clinical selection criteria shown in . At this stage 762 families suspected as having Lynch syndrome were registered at our institute and in 224 families pathogenic germline mutations were identified. Descriptions of the mutation spectrum found in these families, genotype-phenotype correlations and pathology were reported elsewhere Citation[6–8]. Here we report on nine novel germline mutations in ten patients who were recruited by the same selection criteria.
Table I. Definition of selection criteria.
All patients gave written informed consent authorizing data documentation, analysis of tumor tissue for HNPCC characteristics and mutation analysis in MMR genes. The study was approved by the Ethics Committee of the Bonn University Medical Faculty.
Immunohistochemical analysis and microsatellite instability
One patient of each family (index patient) underwent the following diagnostic procedure: tumor material of the index patient was analyzed both for expression of the MMR proteins MLH1, MSH2, MSH6 and PMS2 by immunohistochemical staining and for microsatellite instabilities (MSI). For immunohistochemical analyses mouse anti-human monoclonal antibodies were used (G219-1129 for MSH2, G168-15 for MLH1, 610918 for MSH6 and 556415 for PMS2, BD PharMingen, San Diego, California, USA). Staining procedures and analysis for microsatellite instability (MSI) were performed as previously described Citation[9]. Tumors were classified as MSI-H (high-level MSI) if at least two markers of the reference panel (BAT25, BAT26, D5S346, D2S123, D17S250) exhibited instabilities. Tumors showing no instabilities in these markers were classified as microsatellite-stable (MSS). A second panel of five alternative markers (BAT40, D10S197, D13S153, MYCL1, D18S58) was applied if only one marker of the first reference panel was instable. In this case, the result was interpreted as MSI-H if at least 30% of all markers showed instabilities; otherwise it was regarded as MSI-L (low-level MSI).
Mutation analysis
Based on results of immunohistochemistry, mutation analysis started with MSH2 in cases showing combined loss of MSH2 and MSH6 proteins, and with MLH1 in cases of combined loss of MLH1 and PMS2 proteins in their tumors. Mutation analysis in the MSH6 or PMS2 genes was performed only in those few patients showing an isolated loss of MSH6 or PMS2 proteins. No mutation analysis was conducted if the tumor was MSS or MSI-L and protein expression was normal. Peripheral blood was drawn from all index patients to extract genomic DNA by standard salting out procedure. Search for small germline mutations in MSH2 and MLH1 was performed as previously described Citation[6]. Mutation analysis in MSH6 was performed by direct sequencing of the entire coding region (primers available on request). For detection of large genomic deletions in MSH2, MLH1, MSH6 and PMS2 multiplex ligation-dependent probe amplification (MLPA) was applied by use of SALSA MLPA Kit P003 for MLH1/MSH2 and P008 for MSH6/PMS2 according to the manufacturer's protocol (MRC-Holland, Amsterdam, The Netherlands).
The mutation nomenclature is according to den Dunnen and Antonarakis (2000) Citation[10].
mRNA analysis.
To determine the effect of the mutation MLH1,c.884 + 4A > G on splicing, fresh venous blood samples (2.5 ml) were collected into PAXgene Blood RNA Tubes (PreAnalytiX, QIAGEN, Hilden, Germany) containing RNA stabilizing solution. Total RNA was extracted by use of the PAXgene Blood RNA Kit (QIAGEN) according to the manufacturer's protocol. First strand cDNA was synthesized from 2 to 3 µg of total RNA by random hexamer-primed reverse transcription with the Superscript first strand system for RT-PCR (Invitrogen GmbH) according to the manufacturer's protocol. RT-PCR fragments were obtained according to standard PCR protocols by use of different primers to generate the appropriate fragments (exon 8F 5′-CAAGGAGAGACAGTAGCTGATGTTAGG-3′ and exon 11R 5′-CCACATTCTGGGGACTGATTTC-3′). Aliquots of the RT-PCR products were separated on a 2% agarose gel and visualized with ethidium bromide on an UV imaging system (BIORAD). DNA fragments were excised from the gel and extracted by using the QIAGEN PCR Purification Kit. Part of RT-PCR product containing both the normal and the aberrant fragments were sequenced directly. Changes of splicing efficiencies due to single base substitutions were evaluated by use of the splice prediction program of the BDGP (http://www.fruitfly.org/seq_tools/splice.html).
Results
Here we report on nine novel pathogenic germline mutations in MLH1, MSH2, MSH6 and PMS2 not listed in the database of the International Collaborative Group on HNPCC (http://www.insight-group.org) or in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/search.html) (). Medical and family histories are shown in . Data were obtained from the patients during genetic counselling and/or from medical reports. In all cases the analysis of the index patient's tumor tissue revealed high MSI and a defect in MMR expression pointing to the MMR gene involved.
Table II. Mutations detected in ten index patients.
Table III. Tumor histories and family tumor histories of ten HNPCC patients identified with novel MMR gene mutations.
Three novel mutations were detected in four patients who exhibited loss of MLH1 and PMS2 proteins in their tumors ():
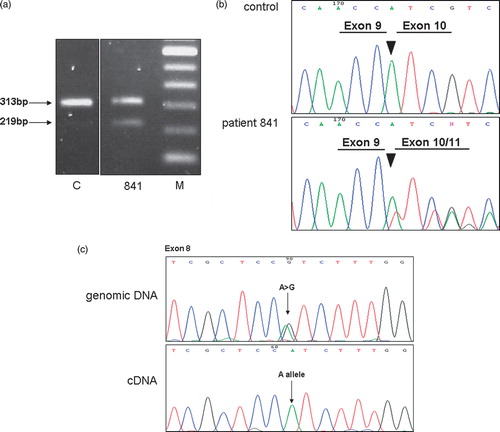
The variant MLH1,c.884 + 4A > G in intron 10 of the MLH1 gene was identified in two apparently unrelated index patients. Due to its position within the not highly conserved position +4 of the splice donor sequence of exon 10 this single base substitution had to be considered as an unclassified variant (UV). This variant segregated with the disease in family 841: the affected sister and the affected father of patient 841 also carry this mutation. According to the splice site prediction program a decrease of splicing efficiency at the splice donor site from 0.93 to 0.59 was predicted for this variant. In order to evaluate the functional relevance of this variant we assessed its influence on splicing by mRNA analysis on index patient 841. RT-PCR products obtained with primers localized in exon 8 and in exon 11, respectively, were separated on an agarose gel and visualized by ethidium bromide staining. In patient 841, a fragment of 219 bp was obtained in addition to the expected fragment of 313 bp (). Sequencing of the 219 bp fragment revealed a loss of exon 10. In order to examine whether the mutant allele was retained in the full length fragment, we made use of the fact that the patient is heterozygous at polymorphic site c.655A > G;p.Val219Ile in exon 8 of the MLH1 gene. The RT-PCR product obtained with a reverse primer localized in exon 10 contained only one allele (A) at nucleotide position 655, suggesting the complete skipping of exon 10 in one allele due to the intronic mutation. The deletion of exon 10 is predicted to result in a frameshift and a premature stop after two codons. We therefore concluded this splice mutation being pathogenic and offered predictive testing for persons at risk in families 841 and 1165.
Figure 1. Characterization of variant MLH1,c.884 + 4A > G. a. Agarose gel showing RT-PCR products obtained in cDNA of index patient 841 and a control (C) by use of a forward primer in exon 8 and a reverse primer in exon 11 of the MLH1 gene. An additional 219 bp fragment due to a deletion of exon 10 was observed in patient 841. DNA ladder with a spacing of 100bp (M). b. Sequencing pattern of the exon 9 – exon 10/11 junction of RT-PCR products of patient 841 showing skipping of exon 10. c. The full-length RT-PCR product of patient 841 contained only one allele (A) at nucleotide position 655, suggesting the complete skipping of exon 10 in one allele due to the intronic mutation.
In addition the frameshift mutations c.1377_1378insA;p.Glu460ArgfsX19 and c.1415_1416delGA;p.Arg472ThrfsX5 in MLH1 were found in patients 1304 and 1289, respectively.
We further identified four novel germline mutations in the MSH2 gene. In these patients tumor tissues showed loss of expression of the MSH2/MSH6 protein complex. The transversions c.301G > T;p.Glu101X, c.842C > A;p.Ser281X, c.859G > T;p.Gly287X and the small deletion c.638_639delTG;p.Leu213GlnfsX18 in MSH2 resulting in a premature stop codon were identified in index patients 1186, 1218, 1197 and 1209, respectively.
The novel nonsense mutation c.2503C > T;p.Gln835X in exon 4 of the MSH6 gene was detected in patient 1097. His colorectal cancer revealed an isolated loss of expression of the MSH6 protein. The patient suffered from testicular cancer at age 25 that is not part of the typical tumor spectrum of Lynch syndrome. Unfortunately the tumor tissue of the testicular cancer was not available for analysis. We performed predictive testing on all five of the patient's siblings. One sister inherited the MSH6 mutation as well and now undergoes the HNPCC surveillance program on a yearly basis.
In patient 908 a selective loss of PMS2 expression pointed to a germline mutation in this gene. A large deletion of exons 1 to 10 of the PMS2 gene was found in this patient. The isolated loss of PMS2 was found in the tumor tissue of his affected sister as well. She is also carrier of the large deletion in the PMS2 gene. Our index patient was at age 32 when his colorectal cancer was diagnosed and his affected sister was at age 42. One older brother died on gastric cancer at age 53; it remains unknown whether he carried the mutation as well. Both affected siblings inherited the mutation from their mother who is 80 years old now. To our knowledge she has never undergone any surveillance program and is healthy until today.
Discussion
We considered the eight novel frameshift, nonsense or large deletion mutations described above as being pathogenic and offer predictive testing to family members at risk.
By mRNA analysis we could demonstrate that the mutation MLH1,c.884 + 4A > G at the less conserved position of the splice donor site in intron 10 leads to a complete deletion of exon 10 in the mutant allele, predicting a frameshift and thus a non-functional MLH1 protein. Therefore we offered predictive testing to the two families with this mutation as well. All mutation carriers now undergo the HNPCC surveillance program on a yearly basis (from the age of 25 onwards but at least 5 years before the onset of the earliest tumor case in the family: annual physical examination, colonoscopy, abdominal sonography, gynaecological examination including transvaginal sonography and gastroscopy from the age of 35 regardless of occurrence of gastric cancer in the family history).
Until now only 14 truncating PMS2 mutations have been described Citation[3], Citation[4], Citation[11–13]. To our knowledge we present the second largest deletion in PMS2 ever described in patients with Lynch syndrome. Hendriks et al. (2006) Citation[4] reported on four genomic rearrangements in PMS2, one of them being a deletion of exon 1–15. Their index patient had four colorectal carcinomas at age 26 and more than 14 adenomas. Unfortunately no more information on other tested family members was given.
It has recently been suggested that mutations in the PMS2 gene do not lead to an autosomal dominant disorder Citation[14] until Worthley et al. (2005) Citation[11] reported on a kindred with autosomal dominant HNPCC due to a familial c.1021delA mutation in the PMS2 gene. Nakagawa et al. (2004) Citation[13] reported on a patient having a deletion of exon 8 of PMS2. The index patient described had a colorectal carcinoma at age 28 but had a healthy father at age 60 who also carries the deletion. In other cases of PMS2 mutations individuals who were heterozygous for a pathogenic PMS2 mutation showed non-penetrance or low penetrance as well Citation[15], Citation[16].
In our family with the deletion of PMS2 exon 1 – 10, a variable penetrance of tumor disease is also noticed.
For investigation of alterations in the MMR genes we have included patients meeting at least one criterion of the Bethesda guidelines Citation[17]. As shown in only two of the ten new families fulfilled the Amsterdam criteria. Thus, the Bethesda criteria combined with examination of tumor tissue perform very well in achieving the intended goal to help guide which families should undergo molecular evaluation for HNPCC. Tumors of all index patients exhibited MSI-H and showed loss of expression of at least one of the MMR proteins. In a previous study on 1 119 unrelated index patients who underwent the standardized screening for MSI-H and IHC a good correlation of these two markers has been described Citation[18]. Further investigations on smaller series confirmed the nearly perfect positive predictive value and specificity as well Citation[19], Citation[20]. The advantage of immunohistochemical analysis is that it may direct mutation analysis because the pattern of staining is specific for the underlying gene defect. The proteins MSH2 and MSH6 bind to form a heterodimer. In absence of MSH2, MSH6 is unstable. Thus, loss of both MSH2 and MSH6 points to a mutation in MSH2. If only MSH6 is absent, the MSH2 protein is still stable and reveals positive staining in immunohistochemistry. Tumors from MSH6 mutation carriers therefore usually show a selective lack of MSH6 staining Citation[4], Citation[21]. Similarly, MLH1 binds with PMS2 to form a heterodimer. Combined loss of MLH1 and PMS2 is characteristic for patients with mutations in MLH1, while an isolated loss of PMS2 protein in tumor tissue points to a germline mutation in PMS2. Our data support these findings as shown in . The identification of the novel germline mutations described underlines the advantage of pre-selection by immunohistochemical staining in tumor tissue for all four MMR proteins in conspicuous families.
Databases
International Collaborative Group on HNPCC: http://www.insight-group.org Human Gene Mutation Database: http://www.hgmd.cf.ac.uk/ac/search.html Splice Site Prediction Program of the Berkeley Drosophila Genome Project (BDGP): http://www.fruitfly.org/seq_tools/splice.html OMIN: #120435; http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=120435 #609310; http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=609310 #600678; http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=600678 #600259; http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=600259
Acknowledgements
The authors thank the patients for their participation in the study and their attending doctors. We thank Marlies Sengteller for excellent technical assistance. The work was supported by a multicenter grant from the German Cancer Aid (Deutsche Krebshilfe e.V. Bonn, project no. 70-2371)
References
- Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med 2003; 348: 919–32
- Plaschke J, Kruger S, Dietmaier W, Gebert J, Sutter C, Mangold E, et al. Eight novel MSH6 germline mutations in patients with familial and nonfamilial colorectal cancer selected by loss of protein expression in tumor tissue. Hum Mut 2004; 23: 285
- Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, et al. Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology 2005; 128: 1160–71
- Hendriks YM, Jagmohan-Changur S, van der Klift HM, Morreau H, van Puijenbroek M, Tops C, et al. Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome). Gastroenterology 2006; 130: 312–22
- Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 1999; 116: 1453–6
- Mangold E, Pagenstecher C, Friedl W, Mathiak M, Buettner R, Engel C, et al. Spectrum and frequencies of mutations in MSH2 and MLH1 identified in 1,721 German families suspected of hereditary nonpolyposis colorectal cancer. Int J Cancer 2005; 116: 692–702
- Pagenstecher C, Wehner M, Friedl W, Rahner N, Aretz S, Friedrichs N, et al. Aberrant splicing in MLH1 and MSH2 due to exonic and intronic variants. Hum Mut 2006; 119: 9–22
- Goecke T, Schulmann K, Engel C, Holinski-Feder E, Pagenstecher C, Schackert HK, et al. Genotype-phenotype comparison of German MLH1 and MSH2 mutation carriers clinically affected with Lynch syndrome: A report by the German HNPCC Consortium. J Clin Oncol 10;24:4285–92.
- Mathiak M, Rutten A, Mangold E, Fischer HP, Ruzicka T, Friedl W, et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol 2002; 26: 338–43
- den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion. Hum Mut 2000; 15: 7–11
- Worthley DL, Walsh MD, Barker M, Ruszkiewicz A, Bennett G, Phillips K, et al. Familial mutations in PMS2 can cause autosomal dominant hereditary nonpolyposis colorectal cancer. Gastroenterology 2005; 128: 1431–6
- Nicolaides NC, Papadopoulos N, Liu B, Wei YF, Carter KC, Ruben SM, et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 1994; 371: 75–80
- Nakagawa H, Lockman JC, Frankel WL, Hampel H, Steenblock K, Burgart LJ, et al. Mismatch repair gene PMS2: Disease-causing germline mutations are frequent in patients whose tumors stain negative for PMS2 protein, but paralogous genes obscure mutation detection and interpretation. Cancer Res 2004; 64: 4721–7
- Gryfe R, Gallinger S. Germline PMS2 mutations: One hit or two?. Gastroenterology 2005; 128: 1506–15
- Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, et al. The molecular basis of Turcot's syndrome. N Engl J Med 1995; 332: 839–47
- Miyaki M, Nishio J, Konishi M, Kikuchi-Yanoshita R, Tanaka K, Muraoka M, et al. Drastic genetic instability of tumours and normal issues in Turcot syndrome. Oncogene 1997; 15: 2877–81
- Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004; 96: 261–8
- Engel C, Forberg J, Holinski-Feder E, Pagenstecher C, Plaschke J, Kloor M, et al. Novel strategy for optimal seqential application of clinical criteria, immunohistochemistry and microsatellite analysis in the diagnosis of hereditary nonpolyposis colorectal cancer. Int J Cancer 2006; 118: 115–22
- Wahlberg SS, Schmeits J, Thomas G, Loda M, Garber J, Syngal S, et al. Evaluation of microsatellite instability and immunohistochemistry for the prediction of germ-line MSH2 and MLH1 mutations in hereditary nonpolyposis colon cancer families. Cancer Res 2002; 62: 3485–92
- Christensen M, Katballe N, Wikman F, Primdahl H, Sorensen FB, Laurberg S, et al. Antibody-based screening for hereditary nonpolyposis colorectal carcinoma compared with microsatellite analysis and sequencing. Cancer 2002; 95: 2422–30
- Fishel R. Signaling mismatch repair in cancer. Nat Med 1999; 5: 1239–41