Abstract
Ionizing radiation is a well established carcinogen for human cells. At low doses, radiation exposure mainly results in generation of double strand breaks (DSBs). Radiation-related DSBs could be directly linked to the formation of chromosomal rearrangements as has been proven for radiation-induced thyroid tumors. Repair of DSBs presumably involves two main pathways, non-homologous end joining (NHEJ) and homologous recombination (HR). A number of known inherited syndromes, such as ataxia telangiectasia, ataxia-telangiectasia like-disorder, radiosensitive severe combined immunodeficiency, Nijmegen breakage syndrome, and LIG4 deficiency are associated with increased radiosensitivity and/or cancer risk. Many of them are caused by mutations in DNA repair genes. Recent studies also suggest that variations in the DNA repair capacity in the general population may influence cancer susceptibility. In this paper, we summarize the current status of DNA repair proteins as potential targets for radiation-induced cancer risk. We will focus on genetic alterations in genes involved in HR- and NHEJ-mediated repair of DSBs, which could influence predisposition to radiation-related cancer and thereby explain interindividual differences in radiosensitivity or radioresistance in a general population.
Ionizing radiation is a well established carcinogen for human cells although the radiation-related cancer is much less frequent compared to that induced by environmental pollutants, tobacco smoking, viruses and food contaminants. Radiation exposure generates a bulk of DNA injuries including numerous base damages, single and double strand breaks (DSBs) Citation[1].
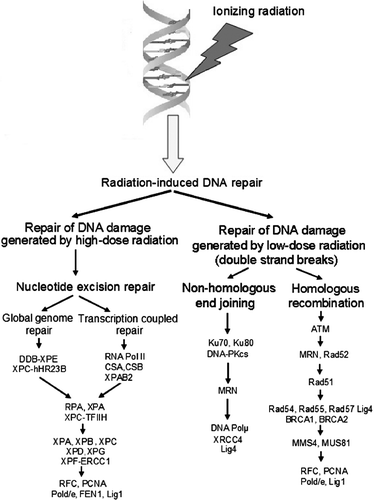
There are several pathways of cellular DNA repair responsible for correction of specific types of DNA damage generated by radiation. Repair of DSBs presumably involves two main mechanisms, non-homologous end joining (NHEJ) and homologous recombination (HR) () Citation[2]. In yeasts, the HR pathway is predominant in DSBs repair, while in vertebrates the NHEJ pathway is believed to be the major mechanism to repair DNA DSBs Citation[3]. Detailed description of the biochemical pathways of DNA repair is beyond the scope of this article as several reviews on the subject have been recently published Citation[4–8].
Figure 1. Radiation exposure at low doses mostly results in DNA double strand breaks (DSBs), which are repaired through homologous recombination and non-homologous end joining. The last pathway is the major mechanism of the repair of DNA DSBs in humans and other mammals.
In human cells, the radiation-induced carcinogenesis is predominantly associated with chromosomal rearrangements. The association has been proven for radiation-induced thyroid tumors Citation[9]. Chromosomal rearrangements, such as Rearranged in Transformation/Papillary Thyroid Carcinomas (RET/PTC), are frequently detected in patients who developed thyroid cancer after the Chernobyl accident Citation[10], Citation[11] or therapeutic irradiation Citation[12].
Radiation-induced DSBs could be directly related to the formation of chromosomal rearrangements Citation[13]. One of the theories explaining the involvement of DNA DSBs into radiation-induced chromosomal rearrangements suggests that the rearrangements are likely to arise from the rejoining of two DSBs located closely in space and time (two-hit mechanism) Citation[14]. According to this theory, a putative mechanism of the rejoining involves the NHEJ pathway of DNA repair. Another theory considers that one radiation-induced DSB is sufficient to initiate an exchange that occurs with an undamaged DNA molecule Citation[15]. In this case, the rearrangement can be generated using HR. However, none of the theories can fully explain all available experimental data on the dose-effect relationship and complexity of radiation-induced aberrations Citation[16].
Analysis of genomic breakpoints in RET/PTC3 rearrangements in post-Chernobyl thyroid tumors showed regions of microhomology composed of 3-5 nucleotides Citation[10], Citation[17], Citation[18]. The modification of sequences at the breakpoints was minimal, typically involving deletion or duplication of 1-3 nucleotides, characteristic of NHEJ. Breakpoints exhibited no particular nucleotide sequence and no recombination-specific motifs. The features of the junction sequences, particularly the high frequency of small terminal deletions, the apparent splicing of DNA ends at microhomologies, and gap-filling on aligned DSBs, are consistent with the known biochemical properties of the classical NHEJ pathway Citation[19]. This provides strong evidence for a dominant role of NHEJ in repair of DSBs and formation of RET/PTC rearrangements after exposure to radiation.
A number of known inherited syndromes are associated with increased radiosensitivity and cancer risk. Many of them are caused by mutations in DNA repair genes Citation[20]. Recent studies also suggest that variations in the DNA repair capacity in the general population may influence cancer susceptibility Citation[21], Citation[22].
In this review, we summarize the current status of DNA repair proteins as potential markers for radiation-induced cancer risk. Since the aberrant repair of radiation-induced DSBs frequently leads to chromosome rearrangements associated with cancer, we will focus on genetic alterations in genes involved into HR- and NHEJ-mediated repair of DSBs, which could influence predisposition to radiation-related cancer and thereby explain interindividual differences in radiosensitivity or radioresistance in a general population.
Genes involved in the homologous recombination pathway of DNA repair
ATM
The ATM gene encodes an important cell cycle checkpoint kinase, a member of the PI3/PI4-kinase family. This enzyme functions as a regulator of a wide variety of downstream proteins, including tumor suppressors p53 and BRCA1, checkpoint kinase CHK2, checkpoint proteins RAD17 and RAD9, and DNA repair proteins NBS1 and SMC1. DNA damage leads to the activation of ATM. This protein kinase phosphorylates and thereby activates SMC1, which is crucial in controlling DNA replication forks and DNA repair after the damage Citation[23]. NBS1 and BRCA1 are required for the recruitment of activated ATM to the sites of DNA breaks followed by phosphorylation of SMC1 by ATM Citation[24].
The ATM-deficient cells are highly sensitive to DNA damage induced by ionizing radiation Citation[25]. These cells are able then to repair the majority of the radiation-induced DSBs with normal kinetics, but fail to repair a subset of breaks irrespective of the initial number of lesions induced. Furthermore, ATM-deficient cells showed no ability to recover following delayed plating after irradiation compared to NHEJ-defective cells Citation[26]. These observations correlate with an extreme cellular sensitivity of patients with ataxia telangiectasia (AT) to ionizing radiation.
Ataxia telangiectasia is an autosomal recessive disorder associated with mutations in the ATM gene. It is characterized by progressive cerebellar ataxia, ocular apraxia, immunodeficiency, chromosomal instability, radiosensitivity and defective cell cycle checkpoint activation Citation[27]. Ataxia telangiectasia patients have a higher risk to develop cancer Citation[28] and obligate heterozygous carriers of ATM mutations may have an increased risk of cancer, particularly breast cancer Citation[29], Citation[30]. Most of population studies failed to show that AT heterozygotes carrying one copy of truncated ATM exhibit significantly increased radiosensitivity Citation[31–36].
Some suggestive evidence was obtained for the association between the ATM codon Asp1853Asn (5557 G > A) single nucleotide polymorphism (SNP) and cancer risk. In France and USA, genetic analysis of breast cancer patients showed strong association between the homozygous carriage of the 5557 G > A variant and high risk of the development of radiotherapy-induced acute skin complications (odds ratio (OR) = 6.76 Citation[37] and 3.1 Citation[38], respectively). This finding was recently confirmed by Andreassen et al. Citation[39] who reported a relationship of the homozygous (AA) and heterozygous (AG) genotypes of the ATM G5557 G > A variant to increased radiosensitivity among Danish breast cancer patients. However, evaluation of the independent cohort of Danish breast cancer patients did not reveal any significant association between this marker and radiosensitivity Citation[40]. For prostate cancer, data on the implication of the ATM codon 1853 polymorphism to radiotherapy-induced complications are controversial. Cesaretti et al. Citation[41] found association between this genetic variant of ATM and the development of radiation-induced proctitis after prostate cancer radiotherapy. However, no relationship was shown between this marker and bladder or rectal toxicity arising from the radiation therapy in Canadian prostate cancer patients Citation[42]. Despite the inconsistence in the association studies, the codon 1853 SNP of ATM seems to play a role in the development of radiotherapy-related complications in breast (and probably prostate) cancer. Additional large-scale population analyses are required for precise confirmation of the involvement of this marker in acute tissue reactions after cancer radiotherapy.
The presence of a large variety of rare missense variants in addition to common polymorphisms in ATM makes it difficult to establish a relationship of this gene to non-radiation and radiation-induced cancer by association studies. However, in those patients who developed cancer after long-term low-dose radiation exposure Citation[36], Citation[43], Citation[44] the frequency of heterozygous carriers of AT missence mutations has been shown to be significantly increased (up to 10%) compared to the general population where the frequency of AT heterozygotes is estimated to be 0.36–1.0% Citation[45].
It has been suggested that missense mutations but not truncating mutations could underline the relationship between the ATM gene and radiosensitivity Citation[46]. Some experimental data support this hypothesis. Angele et al. Citation[37] found that breast cancer individuals heterozygous for both AT mutations IVS22-77 T > C and IVS48 + 238 C > G are significantly more sensitive to radiotherapy (OR = 1.75) than patients who are heterozygous only for one of these mutations. In another study, Angele and coauthors Citation[47] showed that after exposure to ionizing radiation cell cycle progression profile of lymphoblastoid cells carrying both the 3161G and the 2572C (858L) variants of ATM is significantly different from that in cell lines with a wild-type ATM gene. Similarly, cell lines carrying the 2572T > C (F858L) and the 3161C > G (P1054R) ATM variants form more micronuclei than normal cells Citation[48].
The mechanisms by which missense mutations could affect the ATM activity are not fully understood. Of the mutations associated with breast cancer, only two, S2592C and SRI2546del3, produce mutant protein lacking the ATM kinase activity, which also has a dominant negative effect on the wild-type protein. The S2592C substitution could inactivate the kinase function by altering the conformation of ATM or disrupting a potential phosphorylation site in the SSQL sequence Citation[49]. Missense mutations situated in regions of the ATM protein, away from the kinase domain, may decrease the ATM kinase function presumably via the protein-protein interaction with intact ATM molecule and further multimerization Citation[49], Citation[50]. Thus, even low levels of mutant ATM protein have the potential to interfere with ATM function and might in this way contribute to cancer susceptibility Citation[46]. It is likely that a reduced amount of ATM protein in heterozygotes may be responsible for the intermediate sensitivity to radiation.
MRN complex
Three proteins, MRE11, RAD50 and NBS1, participate in the formation of the so called MRN complex. This complex plays a key role in DNA damage detection and activation of the DNA damage response. The MRN complex serves as a flexible link between the ends of broken DNA, and upon binding to damaged DNA, the MRN complex undergoes a series of conformational changes to activate ATM, increase ATM affinity for its substrates Citation[51], and retain active ATM at sites of DNA damage Citation[52].
Mutations in MRE11 could result in deficiency of the MRE11 protein and lead to ataxia-telangiectasia like-disorder (ATLD) Citation[53]. This autosomal recessive disease is characterized by a higher sensitivity to radiation exposure. Delia et al. Citation[54] reported the impaired response to γ-irradiation of lymphoblastoid cells lines and fibroblasts derived from the ATLD patients and those carrying the ATLD-associated mutations 1422C > A (T481K) and 1714C > T (R571X) in MRE11. However, not all ATLD-linked MRE11 mutations are truncating. Fernet et al. Citation[55] found a missense mutation (630G > C, W210C) in the MRE11 gene from Arabic ATLD patients that is located in the nuclease domain of the Mre11 protein and does not affect its expression. Cells homozygous for the 630G > C mutation express normal levels of MRE11 and RAD50 but a very low level of the NBS1 protein, are unable to form the MRE11 foci and show enhanced radiosensitivity Citation[55]. However, to date, the ATLD patients have not been shown to have an increased risk to develop cancer Citation[53].
The observation why ALTD patients are less vulnerable to cancer than those with AT syndrome might be explained by a much broader function of ATM in the cell compared to that of MRE11. The role of MRE11 is focused on the activation of HR-mediated DNA repair while the ATM protein kinase plays a critical role in maintaining genome integrity by activating a biochemical chain reaction that in turn leads to cell cycle checkpoint activation and repair of DNA damage. ATM targets include well-known tumor suppressor genes such as p53 and BRCA1, both of which play an important role in predisposition to breast cancer Citation[56].
Frameshift mutations of the coding nucleotide (T)11 repeat of the RAD50 gene and mutations (484del88) of the microsatellite located in intron 4 of the MRE11 gene leading to the protein truncation have been frequently observed in colorectal and gastrointestinal cancers that developed in patients with deficiency of the DNA mismatch repair (MMR) system Citation[57–59]. MMR-deficient colon cancer cells with mutated RAD50 and MRE11 had impaired expression of both genes, decreased NHEJ repair activity, genome instability and increased sensitivity to γ-irradiation Citation[59]. However, Lefevre et al. Citation[60] found no mutation in the genes constituting the MRN complex in human MMR-stable radiation-induced tumors exhibiting genomic instability.
Mutations in NBS1 are associated with cancer-predisposing Nijmegen breakage syndrome (NBS) that characterized with increased chromosome instability, immunodeficiency and radiosensitivity Citation[61]. Over 90% of patients are homozygous for a founder mutation (657del5): a deletion of five base pairs, which leads to a frameshift and protein truncation. Most of population studies provided evidence for association of this mutation with higher risk of different kinds of cancer, and this risk could be substantial in ethnic groups with high frequency of the founder mutation, for example in Poland and Czech Republic Citation[62–64]. For heterozygous carriers of the 657del5 mutation, the cancer risk tends to increase with age Citation[64].
Several missense mutations such as R215W, S93L, D95N and I171V have been identified in the NBS1 gene in tumor cells of patients with acute lymphoblastic leukaemia Citation[65]. Three of these mutations are located in the FHA or BRCT domains responsible for interaction with ATM and histone H2AX Citation[66]. Inactivating mutations were not found on the second allele in these cells, suggesting that the amino acid substitutions have a dominant negative effect.
In human heterozygotes, the existence of a truncated NBS1 protein produced by alternative translation Citation[67], Citation[68] and capable of interaction with MRE11 would be compatible with a dominant negative mechanism. Studies in human populations with high incidence of NBS found increased incidence of cancer in patients heterozygous for NBS-related mutations. The observed frequency of malignances in heterozygous patients significantly exceeded the expected value Citation[63], Citation[69]. These results suggest that heterozygous carriers of NBS1 mutations may indeed have an enhanced risk to develop malignant tumors, such as melanoma, breast cancer, and colorectal cancer.
Common polymorphisms in NBS1 were evaluated for possible association with a variety of cancers, but consistent positive results have been obtained only for lung cancer: the C allele of the codon 185 dimorphism (E185Q, G > C) was shown to modulate lung cancer risk in several populations Citation[70–73]. This polymorphic marker has been recently tested for possible relation to enhanced risk of radiotherapy-induced acute complications in breast cancer patients in two large-scale population studies involving more then 2 000 cases and controls Citation[74], Citation[75]. Despite that both studies had more than 80% power to detect a 1.9-fold risk in carriers of the NBS1185Q allele no significant association with radiosensitivity was shown. Therefore, it is unlikely that the E185Q SNP of NBS1 could be a major risk factor for clinical radiosensitivity in breast cancer. However, we cannot exclude that this missence mutation could be involved in clinical radiosensitivity as a part of the complex genotype containing susceptibility variants of the major DNA repair genes that have potential to impact increased risk to radiotherapy-induced complications Citation[76].
RAD51 gene family
RAD51 gene family consists of several proteins that show DNA-stimulated ATPase activity and property for preferential binding to single-stranded DNA and forming complexes with each other Citation[77]. RAD51 participates in a common DNA damage response pathway associated with the activation of HR and DSB repair. RAD51 binds to single- and double-stranded DNA and exhibits DNA-dependent ATPase activity. This protein underwinds duplex DNA and forms helical nucleoprotein filaments at the site of DNA break.
Two SNPs, -135 G > C and -172 C > T, have been found in the promoter region of the RAD51 gene Citation[78]. Both are functional and result in increased promoter activity Citation[79]. The + 135 G > C has been found to be associated with predisposition to breast cancer, especially at subgroup of patients with mutations in BRCA2 Citation[80], Citation[81], to ovarian cancer Citation[82], and acute myeloid leukemia Citation[83]. The + 135C allele of RAD51 was reported to be associated with increased risk of radiotherapy-induced acute myeloid leukemia (OR = 2.66) Citation[84]. A synergic interaction between the + 135 G > C SNP of RAD51 and the C/T substitution at the 3′ UTR of the HLX1 homeobox gene, which is important for hematopoietic development, was observed. This resulted in a significant 9.5-fold increase in the risk of acute myeloid anemia in carriers of predisposing variants of both genes Citation[84]. However, Damaraju et al. Citation[42] failed to find significant association between the + 135 G > C promoter polymorphism and risk of development of radiation-induced complications in patients with prostate cancer treated with radiotherapy.
Because a guanine-to-cytosine substitution at position + 135 of the RAD51 is a gain-of-function mutation, it is expected to result in increased activity of RAD51. This effect is opposite to those found for most of the other genetic variations in DNA repair genes, which result in the decrease of function. Interestingly, increased (up to 7-fold) levels of RAD51 have been observed in different tumor cell lines Citation[85]. This finding suggests that up-regulation of RAD51 recombinase may play a role in the increased risk of tumorigenesis Citation[86].
In humans, RAD51 paralogs (RAD51B, RAD51C, RAD51D, XRCC2 and XPCC3) facilitate HR mediated by RAD51 Citation[87]. In chicken DT40 cells, knocking out for any of the RAD51 paralogs results in very similar phenotypes, such as defective HR, chromosome instability, mild sensitivity (only 2-fold higher then that of a wild-type) to γ-irradiation but high sensitivity (8 times higher than in normal cells) to cisplatin, a DNA cross-linking chemotherapeutic agent Citation[88]. Similarity in properties of mutant cells deficient for RAD51 paralogs suggested that these proteins act as a single functional unit during HR.
A polymorphic arginine-to-histidine substitution located at codon 188 (R188H; 31479 G > A) has been found in exon 3 of the XRCC2 gene Citation[89]. XRCC2-deficient DT40 cells transfected with the human XRCC2 R188H variant displayed a more resistant phenotype to cisplatin than wild-type clones due to the restored DNA repair activity of the XRCC2 protein Citation[90]. Genetic studies have shown that the R188H variant of XRCC2 could modulate risk of sporadic breast cancer Citation[89], Citation[91], Citation[92] and epithelial ovarian cancer Citation[93]. However, this SNP failed to show significant relation to radiation-induced complications in patients with BC Citation[74], Citation[75] as well as to bladder/rectal toxicity in radiotherapy-treated subjects with prostate cancer Citation[42].
Another RAD51 paralog, XRCC3 has been more extensively tested for association with acute side effects of radiotherapy in different types of cancer (). A threonine-to-methionine substitution at codon 241 (T241M, 18067 C > T) of XRCC3 has been frequently evaluated in the case-control studies. However, most of the studies failed to show association of this marker with radiotherapy-induced complications such as meningioma Citation[94] and hypersensitivity to ionizing radiation in breast cancer Citation[74], Citation[75], Citation[95–97] and gynecologic tumors Citation[98]. Andreassen et al. Citation[99] reported association between the Thr/Thr241 variant of XRCC3 and enhanced risk of radiation-induced subcutaneous fibrosis in 41 Danish breast cancer patients treated with post-mastectomy radiotherapy, but failed to confirm the results in the independent cohort of 120 post-mastectomy subjects Citation[40].The T241M polymorphism of XRCC3 was found to be associated with radiosensitivity in non-cancer subjects () Citation[100], Citation[101]. The XRCC3 M241T was shown to be associated with several types of non-radiation-induced (sporadic) cancer such as melanoma skin cancer Citation[102], basal cell carcinoma Citation[103], differentiated thyroid cancer Citation[104] and bladder cancer Citation[105].
Table I. Summary of case-control and functional studies for association of different variants of the XRCC3 gene with radiosensitivity
Functional studies showed no significant differences in DNA repair activity between the XRCC3 T241M variant and wild-type protein. Cells having the T241M variant of XRCC3 exhibited the same sensitivity to the interstrand cross-linking agent mytomycin C as those expressing the wild-type protein Citation[106].
These results suggest that the M241T SNP may not be directly associated with radiosensitivity. However, this SNP could be in linkage disequilibrium with another gene (or another genetic variation within XRCC3) responsible for the radiation-induced cancer association. For XRCC3, the association with radiation-induced complications or adverse effects of radiotherapy in cancer was showed for several polymorphic markers including a dinucleotide microsatellite located in intron 3 Citation[107], Citation[108] and two SNPs, 5′ UTR 4541A > G and IVS5-14 A > G Citation[42], Citation[98]. To date, it is unclear whether these markers are functionally significant and hence should be evaluated.
BRCA1 and BRCA2
BRCA1 participates in early steps of DNA repair, playing a role in regulation and promotion of HR. In response to DSBs, BRCA1 is phosphorylated by kinases including ATM, Rad-3 related, and checkpoint kinase 2, and may act in DNA damage-induced signal transduction Citation[109]. Furthermore, BRCA1 is a component of large multiprotein complexes such as BASC (BRCA1-associated genome-surveillance complex) Citation[110], where it can influence the choice of repair pathway utilized depending upon the type of DNA lesion. A specific role for BRCA1 in these complexes might involve regulation of initial DNA DSB processing by the MRN complex Citation[111], which then allows further progression along the HR pathway. BRCA1 is involved in a wide spectrum of other cellular processes such as cell-cycle regulation, transcriptional regulation and chromatin remodelling.
In contrast to BRCA1, BRCA2 functions are largely limited to DNA repair and recombination. BRCA1 and BRCA2 regulate the core HR machinery via control of the RAD51 recombinase. They bind to RAD51 through eight evolutionary conserved binding domains called the BRC repeats Citation[112]. BRCA1 could also bind to single-stranded DNA via the C-terminal domain, the structure of which is critical to the ability of BRCA2 to promote recombination. Following DNA damage and initial DSB processing, BRCA2 relocalizes to the site of DNA damage Citation[113].
Germ-line mutations in BRCA1 and BRCA2 are associated with breast and ovarian cancer. Women heterozygous for the BRCA1/BRCA2 mutations have an elevated risk of developing breast cancer (up to 85% of multi-case families), ovarian cancer and other cancers Citation[114]. However, most studies failed to find a relationship between heterozygous BRCA1/BRCA2 mutations and increased hypersensitivity to radiation in patients with breast and ovarian cancer Citation[115–123].
Observations on the repair of DSBs induced by ionizing radiation in human carcinoma cells deficient in BRCA1 and BRCA2 revealed normal rejoining of DNA DSBs, therefore suggesting for a lack of the direct role of BRCA1 or BRCA2 in the rejoining of radiation-induced DSBs in the genome of human tumor cells Citation[124]. Loss of BRCA function may therefore not substantially sensitize tumors to ionizing radiation.
Evaluation of common polymorphisms within the BRCA1 and BRCA2 genes did not revealed association with radiotherapy-induced toxicity in cancer patients Citation[41], Citation[125]. Although more population studies are required to verify a relationship between the common BRCA1/2 polymorphisms and clinical radiosensitivity in breast and ovarian cancer, the currently available data provide no evidence that genetic alterations in BRCA1/2 play a significant role in predisposition to radiation-induced cancer.
Genes involved in the non-homologous end joining pathway of DNA repair
DNA-dependent protein kinase
DNA-dependent protein kinase (DNA-PK) is a multiprotein complex consisting of the regulatory subunit (Ku heterodimer) and catalytic subunit (DNA protein kinase; DNA-PKcs). Ku heterodimer is comprised from Ku70 and Ku80 subunits that bind to free DNA ends at the break site to keep them in proximity. Then DNA-PKcs binds to the Ku heterodimer forming the DNA-PK complex that stimulates DNA-PKcs activity through the autophosphorylation Citation[126]. This interaction also protects free DNA ends at the site of DSB from nuclease digestion prior to ligation.
DNA-PK is a nuclear serine/threonine kinase, a member of the phosphatidylinositol-3-kinase superfamily. Phosphorylated DNK-PK is active and able to phosphorylate a number of other proteins including those that participate in DNA repair such as Ku70, Ku80, Artemis, XRCC4, replication protein A, Werner syndrome protein, H2AX and several others Citation[127].
M059J cells deficient for DNA-PKcs exhibit a radiosensitive phenotype and are defective in the repair of chromosomal DSBs that reflects a crucial role of this enzyme in maintaining chromosome stability Citation[128]. In DNA-PK-proficient M059K cells, irradiation with low doses of x-rays followed by treatment with wortmannin, an inhibitor of DNA-PK and ATM, results in the recruitment of a slow, error-prone repair process that favored the increased formation of chromosome aberrations Citation[129]. The radiosensitivity of M059J can be complemented by fusion with murine SCID cells harboring human chromosome 8 Citation[130], highlighting the dependence on DNA-PK activity for efficient repair of radiation-induced DNA damage. Down-regulation of DNA-PK using an RNA interference approach also results in significantly enhanced sensitivity to ionizing radiation and DNA-damaging agents Citation[131–134].
Studies on BALB/c mice suggest that genetic alterations within the Prkdc, the mouse ortholog of human DNA-PKcs, could be related to increased radiosensitivity and cancer risk. Two BALB/c strain-specific polymorphisms in the coding region of Prkdc, have been identified Citation[135].The unique PrkdcBALB variant gene carrying the M3844V amino acid substitution in the phosphatidylinositol 3-kinase domain and the R2140C SNP downstream of the putative leucine zipper domain is shown to be associated with decreased DNA-PK catalytic subunit activity and increased susceptibility to radiation-induced genomic instability in primary mammary epithelial cells. In addition, these SNP showed association with radiation-induced apoptosis and lymphomagenesis in mice Citation[136]. These data provide evidence for a possible role of DNA-PK in radiation-induced carcinogenesis. However, no data are available so far on whether genetic variations in DNA-PK can influence predisposition to radiation-induced cancer. Polymorphisms in the human Ku70 and Ku80 genes have been only tested for possible relationship to sporadic breast cancer, but no association has been found Citation[87], Citation[137], Citation[138].
XRCC4/DNA Ligase IV complex
Together with DNA-PK, x-ray repair cross complementing protein 4 (XRCC4) and DNA ligase IV serve as core components of the NHEJ repair complex on DNA ends Citation[139], Citation[140]. XRCC4/DNA ligase IV is able to ligate one strand even when the antiparallel strand can not be ligated as long as at least two base pairs (>4 hydrogen bonds) stabilize the two DNA ends at the overhang. Then, the remaining single-stranded break can be repaired as a single-stranded lesion.
Some evidence suggests that genetic alterations in XRCC4 and ligase IV may promote genomic instability and radiosensitivity. Truncated mutations in XRCC4 result in the deficiency of the protein and radiosensitive phenotype of the respective cell lines Citation[141], Citation[142]. In XRCC4 mutant cell lines, both efficiency and fidelity in repair of DSBs are significantly reduced Citation[143]. The 180BR cell line derived from a radiosensitive leukemia patient is characterized by the R278H mutation resided in the catalytic center of ligase IV that leads to impaired activity of the mutated enzyme Citation[144]. Mutations in human ligase IV are linked to Ligase IV syndrome, a disorder associated with microcephaly, several immunodeficiency, cell radiosensitivity and chromosome instability Citation[145–147]. The clinical phenotype of this syndrome is similar to that of severe combined immunodeficiency (SCID) observed in mice lacking XRCC4, ligase IV or any other NHEJ factor.
To date, it is unclear whether XRCC4 and LIG4 polymorphisms could confer predisposition to radiation-induced tumors in the general population or be associated with radiosensitivity in cancer patients due to the lack of large population data. There are only few genetic studies that aimed to evaluate a possible relationship between genetic alterations in the XRCC4 and LIG4 genes and radiosensitivity in humans. Wilding et al. Citation[148] failed to find association between the I134T XRCC4 variant and translocation frequencies in peripheral blood lymphocytes from cancer-free former workers of British Nuclear Fuels facility at Sellafield. In ligase IV, Borgmann et al. Citation[35] reported a 3012delC mutation in lymphoblastoid cell lines from radiosensitive patients who developed severe side effects after radiotherapy of neck and head cancer. However, heterozygotes for this mutation have also been found in cancer-free controls suggesting that this deletion is a relatively common polymorphism in the general population. The deletion did not affect the coding sequence of the ligase IV gene suggesting that this genetic alteration is most likely functionally neutral Citation[35].
Artemis nuclease and DNA polymerase µ
To aid in the successful repair of DSBs, it has been suggested that NHEJ requires the action of a DNA polymerase. DNA polymerases that could participate in NHEJ include enzymes belonging to the X family that comprises, in addition to DNA polymerase β involved in base-excision repair, polymerases γ,?λ, σ?and µ Citation[149]. DNA polymerase µ (pol µ), a template-dependent polymerase, was shown to participate in radiation-induced DNA repair through the interaction with Ku in a manner dependent on the XRCC4/ligase IV complex Citation[150]. Pol µ also aids the ligation of complementary ends by the XRCC4/ligase IV and Ku complexes. A role of pol λ and pol σ in repair of radiation-induced DSBs is unclear but can not be excluded.
Artemis is a nuclease with 5′–3′ endonuclease activity that removes 5′ overhangs and shortens 3′ overhangs. In vitro phosphorylation of Artemis by DNA-PK activates the hairpin-opening activity of Artemis, which is a prerequisite for V(D)J recombination Citation[151]. This nuclease was found to be a target for ATM-dependent Citation[152] or DNA-PK-mediated Citation[153] phosphorylation after exposure to ionizing radiation.
Irradiated Artemis-deficient human fibroblasts are unable to repair 15–20% of DSBs suggesting that this endonuclease is responsible for processing of a subset of complex DSBs in cells that have no G1 cell cycle checkpoint defects Citation[154]. On the other hand, Artemis cells display at least moderate radiosensitivity Citation[155], Citation[156]. Mutations in this nuclease are associated with a variant of human and murine SCID characterized by poorly developed immune system, radiosensitivity and defect in NHEJ Citation[157–160].
No studies on genetic alterations within pol µ, pol λ and Artemis and their possible association with radiation-induced cancer have been reported to date. However, these genes remain to be promising candidates for biomarkers of clinical radiosensitivity in cancer patients.
Conclusion
To date, significant advances have been achieved in evaluating the role of genetic variations within DNA repair genes in clinical radiosensitivity in cancer. Missense mutations in ATM associated with the AT disease phenotype and truncated mutations in NBS1 associated with Nijmegen breakage syndrome are likely to contribute to increased risk of radiation-induced cancer in general population. Several polymorphisms within the RAD51 and XRCC3 have been suggested to be associated with radiosensitivity in cancer.
Most of case-control studies searching for the contribution of genetic alterations within DNA repair genes to susceptibility to radiation-related cancer have been focused on genes involved in HR. However, since NHEJ is likely to be the major mechanism of repair of radiation-induced DSBs in humans, a role of NHEJ-linked genes in predisposition to radiation-induced cancer remains to be explored in greater detail. Additional efforts are needed to find novel genetic variants of DNA repair genes involved in HR that confer susceptibility to radiation-induced cancer as well as to confirm already discovered disease-associated variants.
Recently, several national and international clinical research projects have been initiated to find markers of genetic predisposition to radiation-induced cancer and clinical radiosensitivity in tumor tissues. National projects include Japanese RadGenomics Citation[161], and Assessment of Polymorphisms for Predicting the Effects of Radiotherapy (RAPPER) and Radiation Complications and Epidemiology (RACE) studies, both of which are UK-based Citation[162]. International projects are presented by the European-based Genetic Pathways for the Prediction of the Effects of Irradiation (GENEPI) Citation[163] and Genetic Predictors of Adverse Radiotherapy (Gene-PARE) Citation[164] studies. In contrast to the GENEPI project involved almost only Caucasians, in the Gene-PARE project, approximately 500 African-Americans will be screened for genetic variants associated with clinical radiosensitivity. The projects are expected to include the evaluation of a variety of candidate genes including DNA repair genes.
To date, the small numbers of individuals showing either an early adverse reaction or a late reaction or both that have been included in many association studies exclude the possibility of addressing whether specific SNPs can influence the temporal aspect of this radiosensitivity. Further association studies in well-characterized large cohorts will be necessary to identify genes that influence the temporal aspects of this adverse response to radiotherapy. However, over the next few years, a considerable molecular characterization of large-scale cohorts of individuals who show therapeutic radiation sensitivity is likely to be achieved. The construction and use of genetic-risk profiles may provide significant improvements in the efficacy of population-based programs of intervention for cancers. This also should help in predicting radiosensitivity that will eventually allow individual tailoring of treatment and reduce the risk of developing acute reactions in anticancer radiotherapy.
Related Research Data
References
- Houtgraaf JH, Versmissen J, van der Giessen WJ. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc Revasc Med 2006; 7: 165–72
- Agarwal S, Tafel AA, Kanaar R. DNA double-strand break repair and chromosome translocations. DNA Repair 2006; 5: 1075–81
- Iliakis G, Wang H, Perrault AR, Boecker W, Rosidi B, Windhofer F, et al. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet Genome Res 2004; 104: 14–20
- Costa RM, Chigancas V, Galhardo Rda S, Carvalho H, Menck CF. The eukaryotic nucleotide excision repair pathway. Biochimie 2003; 85: 1083–99
- Lees-Miller SP, Meek K. Repair of DNA double strand breaks by non-homologous end joining. Biochimie 2003; 85: 1161–73
- Dudas A, Chovanec M. DNA double-strand break repair by homologous recombination. Mutat Res 2004; 566: 131–67
- Sancar A, Lindsey-Boltz LA, Unsal-Kaccmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73: 39–85
- Willers H, Dahm-Daphi J, Powell SN. Repair of radiation damage to DNA. Br J Cancer 2004; 90: 1297–301
- Ciampi R, Nikiforov YE. Alterations of the BRAF gene in thyroid tumors. Endocr Pathol 2005; 16: 163–72
- Nikiforov YE, Koshoffer A, Nikiforova M, Stringer A, Fagin JA. Chromosomal breakpoint positions suggest a direct role for radiation in inducing illegitimate recombination between the ELE1 and RET genes in radiation-induced thyroid carcinomas. Oncogene 1999; 18: 6330–4
- Richter H, Braselmann H, Hieber I, Thomas G, Bogdanova T, Tronko N, et al. Chromosomal imbalances in post-Chernobyl thyroid tumors. Thyroid 2004; 14: 1061–4
- Saenko V, Rogounovitch T, Shimizu-Yoshida Y, Abrosimov A, Lushnikov E, Roumiantsev P, et al. Novel tumorigenic rearrangement, Delta rfp/ret, in a papillary thyroid carcinoma from externally irradiated patient. Mutat Res 2003; 527: 81–90
- Rothkamm K, Huhne M, Jeggo PA, Lobrich M. Radiation-induced genomic rearrangements formed by nonhomologous end-joining of DNA double-strand breaks. Cancer Res 2001; 61: 3886–93
- Savage, Jr. A brief survey of aberration origin theories. Mutat Res 1998; 404: 139–47
- Chadwick KH, Leenhouts HP. The rejoining of DNA double-strand breaks and a model for the formation of chromosomal rearrangements. Int J Radiat Biol Relat Stud Phys Chem Med 1978; 33: 517–29
- Edwards AA. Modelling radiation-induced chromosome aberrations. Int J Radiat Biol 2002; 78: 551–8
- Bongarzone I, Butti MG, Fugazzola L, Pacini F, Pinchera A, Vorontsova TV, et al. Comparison of the breakpoint regions of ELE1 and RET genes involved in the generation of RET/PTC3 oncogene in sporadic and in radiation-associated papillary thyroid carcinomas. Genomics 1997; 42: 252–9
- Klugbauer S, Pfeiffer P, Gassenhuber H, Beimfohr C, Rabes HM. RET rearrangements in radiation-induced papillary thyroid carcinomas: High prevalence of topoisomerase I sites at breakpoints and microhomology-mediated end joining in ELE1 and RET chimeric genes. Genomics 2001; 73: 149–60
- Povirk LF. Biochemical mechanisms of chromosomal translocations resulting from DNA double-strand breaks. DNA Repair (Amst) 2006; 5: 1199–212
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature 2001; 411: 366–74
- Lopez-Cima MF, Gonzalez-Arriada P, Garcia-Castro L, Pascual T, Marron MG, Puente XS, et al. Polymorphisms in XPC, XPD, XRCC1, and XRCC3 DNA repair genes and lung cancer risk in a population of northern Spain. BMC Cancer 2007; 7: 162
- Bau DT, Mau YC, Ding SL, Wu PE, Shen CY. DNA double-strand break repair capacity and risk of breast cancer. Carcinogenesis 2007; 28: 1726–30
- Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev 2004; 18: 1423–38
- Kitagawa R, Kastan MB. The ATM-dependent DNA damage signaling pathway. Cold Spring Harb Symp Quant Biol 2005; 70: 99–109
- Morrison C, Sonoda E, Takao N, Shinohara A, Yamamoto K, Takeda S. The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J 2000; 19: 463–71
- Kuhne M, Riballo E, Rief N, Rothkamm K, Jeggo PA, Lobrich M. A double-strand break repair defect in ATM-deficient cells contributes to radiosensitivity. Cancer Res 2004; 64: 500–8
- Auerbach AD, Veralnder PC. Disorders of DNA replication and repair. Curr Opin Pediatr 1997; 9: 600–16
- Khanna KK. Cancer risk and the ATM gene: A continuing debate. J Natl Cancer Inst 2000; 92: 795–802
- Broeks A, Urbanus JH, Floore AN, Dahler EC, Klijn JG, Rutgers EJ, et al. ATM-heterozygous germline mutations contribute to breast cancer-susceptibility. Am J Hum Genet 2000; 66: 494–500
- Thorstenson YR, Roxas A, Kroiss R, Jenkins MA, Yu KM, Bachrich T, et al. Contributions of ATM mutations to familial breast and ovarian cancer. Cancer Res 2003; 63: 3325–33
- Appleby JM, Barber JB, Levine E, Varley JM, Taylor AM, Stankovic T, et al. Absence of mutations in the ATM gene in breast cancer patients with severe responses to radiotherapy. Br J Cancer 1997; 76: 1546–9
- Hall EJ, Schiff PB, Hanks GE, Brenner DJ, Russo J, Chen J, et al. A preliminary report: Frequency of A-T heterozygotes among prostate cancer patients with severe late responses to radiation therapy. Cancer J Sci Am 1998; 4: 385–9
- Weissberg JB, Huang DD, Swift M. Radiosensitivity of normal tissues in ataxia-telangiectasia heterozygotes. Int J Radiat Oncol Biol Phys 1998; 42: 1133–6
- Oppitz U, Bernthaler U, Schindler D, Sobeck A, Hoehn H, Platzer M, et al. Sequence analysis of the ATM gene in 20 patients with RTOG grade 3 or 4 acute and/or late tissue radiation side effects. Int J Radiat Oncol Biol Phys 1999; 44: 981–8
- Borgmann K, Roper B, El Awady R, Brackrock S, Bigalke M, Dork T, et al. Indicators of late normal tissue response after radiotherapy for head and neck cancer: Fibroblasts, lymphocytes, genetics, DNA repair, and chromosome aberrations. Radiother Oncol 2002; 64: 141–52
- Bremer M, Klopper K, Yamini P, Bendix-Waltes R, Dork T, Karstens JH. Clinical radiosensitivity in breast cancer patients carrying pathogenic ATM gene mutations: No observation of increased radiation-induced acute or late effects. Radiother Oncol 2003; 69: 155–60
- Angele S, Romestaing P, Moullan N, Vuillaume M, Chapot B, Friesen M, et al. ATM haplotypes and cellular response to DNA damage: Association with breast cancer risk and clinical radiosensitivity. Cancer Res 2003; 63: 8717–25
- Ho AY, Fan G, Atencio DP, Green S, Formetti SC, Haffty BG, et al. Possession of ATM sequence variants as predictor for late normal tissue responses in breast cancer patients treated with radiotherapy. Int J Radiat Oncol Biol Phys 2007; 69: 677–84
- Andreassen CN, Overgaard J, Alsner J, Overgaard M, Herskind C, Cesaretti JA, et al. ATM sequence variants and risk of radiation-induced subcutaneous fibrosis after postmastectomy radiotherapy. Int J Radiat Oncol Biol Phys 2006; 64: 776–83
- Andreassen CN, Alsner J, Overgaard M, Sorensen FB, Overgaard J. Risk of radiation-induced subcutaneous fibrosis in relation to single nucleotide polymorphisms in TGFB1, SOD2, XRCC1, XRCC3, APEX and ATM – a study based on DNA from formalin fixed paraffin embedded tissue samples. Int J Radiat Biol 2006; 82: 577–86
- Cesaretti JA, Stock RG, Atencio DP, Peters SA, Peters CA, Burri RJ, et al. A genetically determined dose-volume histogram predicts for rectal bleeding among patients treated with prostate brachytherapy. Int J Radiat Oncol Biol Phys 2007; 68: 1410–6
- Damaraju S, Murray D, Dufour J, Carandang D, Myrehaug S, Fallone G, et al. Association of DNA repair and steroid metabolism gene polymorphisms with clinical late toxicity in patients treated with conformal radiotherapy for prostate cancer. Clin Cancer Res 2006; 12: 2545–54
- Iannuzzi CM, Atencio DP, Green S, Stock RG, Rosenstein BS. ATM mutations in female breast cancer patients predict for an increase in radiation-induced late effects. Int J Radiat Oncol Biol Phys 2002; 52: 606–13
- Schneider J, Philipp M, Yamini P, Dork T, Woitowitz HJ. ATM gene mutations in former uranium miners of SDAG Wismut: A pilot study. Oncol Rep 2007; 17: 477–82
- Fernet M, Hall J. Genetic biomarker of therapeutic radiation sensitivity. DNA Repair (Amst) 2004; 3: 1237–43
- Gatti RA, Tward A, Concannon P. Cancer risk in ATM heterozygotes: A model of phenotypic and mechanistic differences between missense and truncating mutations. Mol Genet Metab 1999; 68: 419–23
- Angele S, Falconer A, Edwards SM, Dork T, Bremer M, Moullan N, et al. ATM polymorphisms as risk factors for prostate cancer development. Br J Cancer 2004; 91: 783–7
- Gutierrez-Enriquez S, Ferenet M, Dork T, Bremer M, Lauge A, Stoppa-Lyonnet D, et al. Functional consequences of ATM sequence variants for chromosomal radiosensitivity. Genes Chromosome Cancer 2004; 40: 109–19
- Scott SP, Bendix R, Chen P, Clark R, Dork T, Lavin MF. Missense mutations but not allelic variants alter the function of ATM by dominant interference in patients with breast cancer. Proc Natl Acad Sci USA 2002; 99: 925–30
- Smith GC, Cary RB, Lakin ND, Hann BC, Teo SH, Chen DJ, et al. Purification and DNA binding properties of the ataxia-telangiectasia gene product ATM. Proc Natl Acad Sci USA 1999; 96: 11134–9
- Paull TT, Lee JH. The Mre11/Rad50/Nbs1 complex and its role as a DNA double-strand break sensor for ATM. Cell Cycle 2005; 4: 737–40
- You Z, Chahwan C, Bailis J, Hunter T, Russell P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol 2005; 25: 5363–79
- Taylor AM, Groom A, Byrd PJ. Ataxia-telangiectasia-like disorder (ATLD) – its clinical presentation and molecular basis. DNA Repair (Amst) 2004; 3: 1219–25
- Delia D, Piane M, Buscemi G, Savio C, Palmeri S, Lulli P, et al. MRE11 mutations and impaired ATM-dependent responses in an Italian family with ataxia-telangiectasia-like disorder. Hum Mol Genet 2004; 13: 2155–63
- Fernet M, Gribaa M, Salih MA, Seidahmed MZ, Hall J, Koenig M. Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder. Hum Mol Genet 2005; 14: 307–18
- Ahmed M, Rahman N. ATM and breast cancer susceptibility. Oncogene 2006; 25: 5906–11
- Kim NG, Choi YR, Baek MJ, Kim YH, Kang H, Kim NK, et al. Frameshift mutations at coding mononucleotide repeats of the hRAD50 gene in gastrointestinal carcinomas with microsatellite instability. Cancer Res 2001; 61: 36–8
- Giannini G, Ristori E, Cerionoli F, Rinaldi C, Zani M, Viel A, et al. Human MRE11 is inactivated in mismatch repair-deficient cancers. EMBO Rep 2002; 3: 248–54
- Koh KH, Kang HJ, Li LS, Kim NG, You KT, Yang E, et al. Impaired nonhomologous end-joining in mismatch repair-deficient colon carcinomas. Lab Invest 2005; 85: 1130–8
- Lefevre SH, Coquelle A, Gonin-Laurent N, Cor A, Vogt N, Chauveninc L, et al. Non-homologous end-joining genes are not inactivated in human radiation-induced sarcomas with genomic instability. J Radiat Res (Tokyo) 2005; 46: 225–31
- Diqweed M, Sperling K. Nijmegen breakage syndrome: Clinical manifestation of defective response to DNA double-strand breaks. DNA Repair (Amst) 2004; 3: 1207–17
- Cybulski C, Gorski B, Debniak T, Gliniewicz B, Mierzejewski M, Masoic B, et al. NBS1 is a prostate cancer susceptibility gene. Cancer Res 2004; 64: 1215–9
- Steffen J, Varon R, Mosor M, Maneva G, Maurer M, Stumm M, et al. Increased cancer risk of heterozygotes with NBS1 germline mutations in Poland. Int J Cancer 2004; 111: 67–71
- Steffen J, Nowakowska D, Niwinska A, Kluska A, Piatkowska M, Wiesnewska A, et al. Germline mutations 657del5 of the NBS1 gene contribute significantly to the incidence of breast cancer in Central Poland. Int J Cancer 2006; 119: 472–5
- Varon R, Reis A, Henze G, von Einsiedel HG, Sperling K, Seeger K. Mutations in the Nijmegen breakage syndrome gene (NBS1) in childhood acute lymphoblastic leukemia (ALL). Cancer Res 2001; 61: 3570–2
- Kobayashi J, Tauchi H, Sakamoto S, Nakamura A, Morishima K, Matsuura S. NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr Biol 2002; 12: 1846–51
- Maser RS, Zinkel R, Petrini JH. An alternative mode of translation permits production of a variant NBS1 protein from the common Nijmegen breakage syndrome allele. Nat Genet 2001; 27: 417–21
- Kruger L, Demuth I, Naitzel H, Varon R, Sperling K, Chrzanowska KH, et al. Cancer incidence in Nijmegen breakage syndrome is modulated by the amount of a variant NBS protein. Carcinogenesis 2007; 28: 107–11
- Seemanova E, Jarolim P, Varon R, Pelz J, Sperling K. Cancer risk in NBS heterozygotes from the Czech Republic. Am J Hum Genet 2002; 71 (Suppl): 238A
- Medina PP, Ahrendt SA, Pollan M, Fernandez P, Sidransky D, Sanchez-Cespedes M. Screening of homologous recombination gene polymorphisms in lung cancer patients reveals an association of the NBS1-185Gln variant and p53 gene mutations. Cancer Epidemiol Biomarker Prev 2003; 12: 699–704
- Lan Q, Shen M, Berndt SI, Bonner MR, He X, Yeager M, et al. Smoky coal exposure, NBS1 polymorphisms, p53 protein accumulation, and lung cancer risk in Xuan Wei, China. Lung Cancer 2005; 49: 317–23
- Ryk C, Kumar R, Thirumaran RK, Hou SM. Polymorphisms in the DNA repair genes XRCC1, APEX1, XRCC3 and NBS1, and the risk for lung cancer in never- and ever-smokers. Lung Cancer 2006; 54: 285–92
- Zienolddiny S, Campa D, Lind H, Ryberg D, Skaug V, Stangeland L, et al. Polymorphisms of DNA repair genes and risk of non-small cell lung cancer. Carcinogenesis 2006; 27: 560–7
- Millikan RC, Player JS, Decotret AR, Tse CK, Keku T. Polymorphisms in DNA repair genes, medical exposure to ionizing radiation, and breast cancer risk. Cancer Epidemiol Biomarker Prev 2005; 14: 2326–34
- Popanda O, Tan XL, Ambrosone CB, Kropp S, Helmbold I, von Fournier D, et al. Genetic polymorphisms in the DNA double-strand break repair genes XRCC3, XRCC2, and NBS1 are not associated with acute side effects of radiotherapy in breast cancer patients. Cancer Epidemiol Biomerker Prev 2006; 15: 2048–50
- Mohrenweiser H, Wilson D, Jones I. Challenges and complexities in estimating both the functional impact and the disease risk associated with the extensive genetic variation in human DNA repair genes. Mutat Res 2003; 526: 93–125
- Thaker J. The RAD51 gene family, genetic instability and cancer. Cancer Lett 2005; 219: 125–35
- Wang WW, Spurdle AB, Kolachana P, Bove B, Modan B, Ebbers SM, et al. A single nucleotide polymorphism in the 5′ untranslated region of RAD51 and risk of cancer among BRCA1/2 mutation carriers. Cancer Epidemiol Biomarkers Prev 2001; 10: 955–60
- Hasselbach L, Haase S, Fischer D, Kolberg HC, Sturzbecher HW. Characterisation of the promoter region of the human DNA-repair gene Rad51. Eur J Gynaecol Oncol 2005; 26: 589–98
- Levi-Lahad E, Lahad A, Eisenberg S, Dagan E, Paperna T, Kasinets L, et al. A single nucleotide polymorphism in the RAD51 gene modifies cancer risk in BRCA2 but not BRCA1 carriers. Proc Natl Acad Sci USA 2001; 98: 3232–6
- Kadouri L, Kote-Jarai Z, Hubert A, Durocher F, Abeliovich D, Glaser B, et al. A single-nucleotide polymorphism in the RAD51 gene modifies breast cancer risk in BRCA2 carriers, but not in BRCA1 carriers or noncarriers. Br J Cancer 2004; 90: 2002–5
- Webb PM, Hopper JL, Newman B, Chen X, Kelemen L, Giles GG, et al. Double-strand break repair gene polymorphisms and risk of breast or ovarian cancer. Cancer Epidemiol Biomarkers Prev 2005; 14: 319–23
- Seedhouse C, Faulkner R, Ashraf N, Das-Gupta E, Russell N. Polymorphisms in genes involved in homologous recombination repair interact to increase the risk of developing acute myeloid leukemia. Clin Cancer Res 2004; 10: 2675–80
- Jawad M, Seedhouse CH, Russell N, Plumb M. Polymorphisms in human homeobox HLX1 and DNA repair RAD51 genes increase the risk of therapy-related acute myeloid leukemia. Blood 2006; 108: 3916–8
- Fan R, Kumaravel TS, Jalali F, Marrano P, Squire JA, Bristow RG. Defective DNA strand break repair after DNA damage in prostate cancer cells: Implications for genetic instability and prostate cancer progression. Cancer Res 2004; 64: 8526–33
- Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T. Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res 2002; 62: 219–25
- Kawabata M, Kawabata T, Nishibori M. Role of recA/RAD51 family proteins in mammals. Acta Med Okayama 2005; 59: 1–9
- Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, et al. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol 2001; 21: 2858–66
- Kuschel B, Auranen A, McBride S, Novik KL, Antoniou A, Lipscombe JM, et al. Variants in DNA double-strand break repair genes and breast cancer susceptibility. Hum Mol Genet 2002; 11: 1399–407
- Danoy P, Sonoda E, Lathrop M, Takeda S, Matsuda F. A naturally occurring genetic variant of human XRCC2 (R188H) confers increased resistance to cisplatin-induced DNA damage. Biochem Biophys Res Commun 2007; 352: 763–8
- Han J, Hankinson SE, Ranu H, De Vivo I, Hunter DJ. Polymorphisms in DNA double-strand break repair genes and breast cancer risk in the Nurses' Health Study. Carcinogenesis 2004; 25: 189–95
- Garcia-Glosas M, Egan KM, Newcomb PA, Brinton LA, Titus-Ernstoff L, Chanock S, et al. Polymorphisms in DNA double-strand break repair genes and risk of breast cancer: Two population-based studies in USA and Poland, and meta-analyses. Hum Genet 2006; 119: 376–88
- Auranen A, Song H, Waterfall C, Dicioccio RA, Kuschel B, Kjaer SK, et al. Polymorphisms in DNA repair genes and epithelial ovarian cancer risk. Int J Cancer 2005; 117: 611–8
- Sadetzki S, Flint-Richter P, Starinsky S, Novikov I, Lerman Y, Goldman B, et al. Genotyping of patients with sporadic and radiation-associated meningiomas. Cancer Epidemiol Biomarker Prev 2005; 14: 969–76
- Hu JJ, Smith RR, Miller MS, Monhrenwaiser HW, Golden A, Case LD. Amino acid substitution variants of APE1 and XRCC1 genes associated with ionizing radiation sensitivity. Carcinogenesis 2001; 22: 917–22
- Hu JJ, Smith TR, Miller MS, Lohman K, Case LD. Genetic regulation of ionizing radiation sensitivity and breast cancer risk. Environ Mol Mutagen 2002; 39: 208–15
- Andreassen CM, Alsner J, Overgaard J, Herskind C, Haviland J, Owen R, et al. TGFB1 polymorphisms are associated with risk of late normal tissue complications in the breast after radiotherapy for early breast cancer. Radiother Oncol 2005; 75: 18–21
- De Ruyck K, Van Eijkeren M, Claes K, Morthier R, De Paepe A, Vral A, et al. Radiation-induced damage to normal tissues after radiotherapy in patients treated for gynecologic tumors: association with single nucleotide polymorphisms in XRCC1, XRCC3, and OGG1 genes and in vitro chromosomal radiosensitivity in lymphocytes. Int J Radiat Oncol Biol Phys 2005; 62: 1140–9
- Andreassen CM, Alsner J, Overgaard M, Overgaard J. Prediction of normal tissue radiosensitivity from polymorphisms in candidate genes. Radiother Oncol 2003; 69: 127–35
- Au WW, Salama SA, Sierra-Torres CH. Functional characterization of polymorphisms in DNA repair genes using cytogenetic challenge assays. Environ Health Perspect 2003; 111: 1843–50
- Aka P, Mateuca R, Buchet IP, Thierens H, Kirsch-Volders M. Are genetic polymorphisms in OGG1, XRCC1 and XRCC3 genes predictive for the DNA strand break repair phenotype and genotoxicity in workers exposed to low dose ionising radiations?. Mutat Res 2004; 556: 169–71
- Winsley SL, Haldar NA, Marsh HP, Bunce M, Marshall SE, Harris AL, et al. A variant within the DNA repair gene XRCC3 is associated with the development of melanoma skin cancer. Cancer Res 2000; 60: 5612–6
- Thirumaran RK, Bermejo JL, Rudnaj P, Gurzau E, Koppova K, Goessler W, et al. Single nucleotide polymorphisms in DNA repair genes and basal cell carcinoma of skin. Carcinogenesis 2006; 27: 1676–81
- Sturgis EM, Zhao C, Zheng R, Wei Q. Radiation response genotype and risk of differentiated thyroid cancer: A case-control analysis. Laryngoscope 2005; 115: 938–45
- Matullo G, Guarrera S, Carturan S, Peluso M, Malaveille C, Davico L, et al. DNA repair gene polymorphisms, bulky DNA adducts in white blood cells and bladder cancer in a case-control study. Int J Cancer 2001; 92: 562–7
- Araujo FD, Pierce AJ, Starkl JM, Jasin M. Variant XRCC3 implicated in cancer is functional in homology-directed repair of double-strand breaks. Oncogene 2002; 21: 4176–80
- Price EA, Bourne SL, Radbourne R, Lawton PA, Lamerdin J, Thompson LH, et al. Rare microsatellite polymorphisms in the DNA repair genes XRCC1, XRCC3 and XRCC5 associated with cancer in patients of varying radiosensitivity. Somat Cell Mol Genet 1997; 23: 237–47
- De Ruyck K, Widing CS, Van Eijkeren M, Morthier R, Tawn EJ, Thierens H. Microsatellite polymorphisms in DNA repair genes XRCC1, XRCC3 and XRCC5 in patients with gynecological tumors: Association with late clinical radiosensitivity and cancer incidence. Radiat Res 2005; 164: 237–44
- Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 1999; 286: 1162–6
- Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev 2000; 14: 927–39
- Zhong Q, Chen CF, Li S, Chen Y, Wang CC, Xiao J, et al. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science 1999; 285: 747–50
- Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, et al. Insights into DNA recombination from the structure of a RAD51-BRCA2 complex. Nature 2002; 420: 287–93
- Yu DS, Sonoda E, Takeda S, Huang CL, Pellegrini L, Blundell TL, et al. Dynamic control of Rad51 recombinase by self-association and interaction with BRCA2. Mol Cell 2003; 12: 1029–41
- Honrado E, Beniztez J, Palacios J. The molecular pathology of hereditary breast cancer: Genetic testing and therapeutic implications. Mol Pathol 2005; 18: 1305–20
- Gaffney DK, Brohet RM, Lewis CM, Holden JA, Byus SS, Neuhausen SL, et al. Response to radiation therapy and prognosis in breast cancer patients with BRCA1 and BRCA2 mutations. Radiother Oncol 1998; 47: 129–36
- Pierce LJ, Strawderman M, Narod SA, Oliviotto I, Eisen A, Dawson L, et al. Effect of radiotherapy after breast-conserving treatment in women with breast cancer and germline BRCA1/2 mutations. J Clin Oncol 2000; 18: 3360–9
- Leong T, Whitty J, Keilar M, Mifsud S, Ramsay J, Birrel G, et al. Mutation analysis of BRCA1 and BRCA2 cancer predisposition genes in radiation hypersensitive cancer patients. Int J Radiat Oncol Biol Phys 2000; 48: 959–65
- Buchholz TA, Wu X, Hussain A, Tucker SL, Mills GB, Haffty B, et al. Evidence of haplotype insufficiency in human cells containing a germline mutation in BRCA1 or BRCA2. Int J Cancer 2002; 97: 557–61
- Nieuewehhuis B, Van Assen-Bolt AJ, Van Waarde-Verhagen MA, Sijmons RH, Van der Hout AH, Bauch T, et al. BRCA1 and BRCA2 heterozygosity and repair of X-ray-induced DNA damage. Int J Radiat Biol 2002; 78: 285–95
- Trenz K, Rothfuss A, Schutz P, Speit G. Mutagen sensitivity of peripheral blood from women carrying a BRCA1 or BRCA2 mutation. Mutat Res 2002; 500: 89–96
- Trenz K, Schutz P, Speit G. Radiosensitivity of lymphoblastoid cell lines with a heterozygous BRCA1 mutation is not detected by the comet assay and pulsed field gel electrophoresis. Mutagenesis 2005; 20: 131–7
- Baeyens A, Thierens H, Claes K, Poppe B, de Ridder L, Vral A. Chromosomal radiosensitivity in BRCA1 and BRCA2 mutation carriers. Int J Radiat Biol 2004; 80: 745–56
- Kirova YM, Stoppa-Lyonnet D, Savignoni A, Sigal-Zafrani B, Fabre N, Fourquet A. Risk of breast cancer recurrence and contralateral breast cancer in relation to BRCA1 and BRCA2 mutation status following breast-conserving surgery and radiotherapy. Eur J Cancer 2005; 41: 2304–11
- Wang H, Zeng ZC, Bui TA, DiBiase SJ, Qin W, Xia F, et al. Nonhomologous end-joining of ionizing radiation-induced DNA double-stranded breaks in human tumor cells deficient in BRCA1 or BRCA2. Cancer Res 2001; 61: 270–7
- Zhang C, Naftalis E, Euhus D. Carcinogen-induced DNA double strand break repair in sporadic breast cancer. J Surg Res 2006; 135: 120–8
- Chan DW, Chen BP, Prithivarajsingh S, Kurimasa A, Story MD, Qin J, et al. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev 2002; 16: 2333–8
- Collis SJ, De Weese TL, Jeggo PA, Parker AR. The life and death of DNA-PK. Oncogene 2005; 24: 949–61
- Lieber MR, Ma Y, Pannicke U, Schwartz K. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 2003; 4: 712–20
- Virsik-Kopp P, Rave-Frank M, Hofman-Huther H, Schmidberger H. Role of DNA-PK in the process of aberration formation as studied in irradiated human glioblastoma cell lines M059K and M059J. Int J Radiat Biol 2003; 79: 61–8
- Hoppe BS, Jensen RB, Kirchgessner CU. Complementation of the radiosensitive M059J cell line. Radiat Res 2000; 153: 125–30
- Peng Y, Zhang Q, Nagasawa H, Okayasu R, Liber HL, Bedford JS. Silencing expression of the catalytic subunit of DNA-dependent protein kinase by small interfering RNA sensitizes human cells for radiation-induced chromosome damage, cell killing, and mutation. Cancer Res 2002; 62: 6400–4
- Collis SJ, Swartz MJ, Nelson WG, De Weese TL. Enhanced radiation and chemotherapy-mediated cell killing of human cancer cells by small inhibitory RNA silencing of DNA repair factors. Cancer Res 2003; 63: 1550–4
- Choudhury A, Cuddihy A, Bristow RG. Radiation and new molecular agents part I: Targeting ATM-ATR checkpoints, DNA repair, and the proteasome. Semin Radiat Oncol 2006; 16: 51–8
- Zhang Y, Zhou J, Cao X, Zhang Q, Lim CU, Bailey SM, et al. Partial deficiency of DNA-PKcs increases ionizing radiation-induced mutagenesis and telomere instability in human cells. Cancer Lett 2007; 250: 63–73
- Yu Y, Okayashi R, Weil MM, Silver A, McCarthy M, Zabriskie R, et al. Elevated breast cancer risk in irradiated BALB/c mice associates with unique functional polymorphism of the Prkdc (DNA-dependent protein kinase catalytic subunit) gene. Cancer Res 2001; 61: 1820–4
- Mori N, Matsumoto Y, Okumoto M, Suzuki N, Yamate J. Variations in Prkdc encoding the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) and susceptibility to radiation-induced apoptosis and lymphomagenesis. Oncogene 2001; 20: 3609–19
- Goode EL, Dunning AM, Kuschel B, Healey CS, Day NE, Ponder BA, et al. Effect of germ-line genetic variation on breast cancer survival in a population-based study. Cancer Res 2002; 62: 3052–7
- Fu YP, Ju JC, Cheng TC, Lou MA, Hsu GC, Wu CY, et al. Breast cancer risk associated with genotypic polymorphism of the nonhomologous end-joining genes: a multigenic study on cancer susceptibility. Cancer Res 2003; 63: 2440–6
- Grawunder U, Wilm M, Wu X, Kulesza P, Wilson TE, Mann M, et al. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature 1997; 388: 492–5
- Drouet J, Delteil C, Lefrancois J, Concannon P, Salles B, Calsou P. DNA-dependent protein kinase and XRCC4-DNA ligase IV mobilization in the cell in response to DNA double strand breaks. J Biol Chem 2005; 280: 7060–9
- Mori M, Itsukaichi H, Nakamura A, Sato K. Molecular characterization of ionizing radiation-hypersensitive mutant M10 cells. Mutat Res 2001; 487: 85–92
- Itsukaichi H, Mori M, Nakamura A, Sato K. Identification of a new G-to-A transition mutation at nucleotide position 129 of the Xrcc4 gene in ionizing radiation-hypersensitive mutant LX830 cells. J Radiat Res (Tokyo) 2003; 44: 353–8
- van Heemst D, Brugmans L, Verkaik NS, van Gent DC. End-joining of blunt DNA double-strand breaks in mammalian fibroblasts is precise and requires DNA-PK and XRCC4. DNA Repair (Amst) 2004; 3: 43–50
- Riballo E, Critchlow SE, Teo SH, Doherty AJ, Priestley A, Broughton B, et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr Biol 1999; 9: 699–702
- O'Driscoll M, Gennery AR, Seidel J, Concannon P, Jeggo PA. An overview of three new disorders associated with genetic instability: LIG4 syndrome, RS-SCID and ATR-Seckel syndrome. DNA Repair (Amst) 2004; 3: 1227–35
- Buck D, Moshous D, de Chasseval R, Ma Y, le Deist F, Cayazzana-Calvo M, et al. Severe combined immunodeficiency and microcephaly in siblings with hypomorphic mutations in DNA ligase IV. Eur J Immunol 2006; 36: 224–35
- Enders A, Fisch P, Schwarz K, Duffner U, Pannicke U, Nikopoulos E, et al. Severe form of human combined immunodeficiency due to mutations in DNA ligase IV. J Immunol 2006; 176: 5060–8
- Wilding CS, Relton CL, Rees GS, Tarone RE, Whitehouse CA, Tawn EJ. DNA repair gene polymorphisms in relation to chromosome aberration frequencies in retired radiation workers. Mutat Res 2005; 570: 137–45
- Ramadan K, Shevelev I, Hubscher U. The DNA-polymerase-X family: Controllers of DNA quality?. Nat Rev Mol Cell Biol 2004; 5: 1038–43
- Mahajan KN, Nick McElhinny SA, Mitchell BS, Ramsden DA. Association of DNA polymerase mu (pol mu) with Ku and ligase IV: Role for pol mu in end-joining double-strand break repair. Mol Cell Biol 2002; 22: 5194–202
- Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 2002; 108: 781–94
- Zhang X, Succi J, Feng Z, Prithiyirajsingh S, Story MD, Legerski RJ. Artemis is a phosphorylation target of ATM and ATR and is involved in the G2/M DNA damage checkpoint response. Mol Cell Biol 2004; 24: 9207–20
- Soubeyrand S, Pope L, De Chasseval R, Gosselin D, Dong F, de Villartay JP, et al. Artemis phosphorylated by DNA-dependent protein kinase associates preferentially with discrete regions of chromatin. J Mol Biol 2006; 358: 1200–11
- Darroudi F, Wiegant W, Meijers M, Friedl AA, van der Burg, Fomina J, et al. Role of Artemis in DSB repair and guarding chromosomal stability following exposure to ionizing radiation at different stages of cell cycle. Mutat Res 2007; 615: 111–24
- Rooney S, Alt FW, Lombard D, Whitlow S, Eckersdorff M, Fleming J, et al. Defective DNA repair and increased genomic instability in Artemis-deficient murine cells. J Exp Med 2003; 197: 553–65
- Musio A, Marrella V, Sobacchi C, Rucci F, Fariselli L, Giliani S, et al. Damaging-agent sensitivity of Artemis-deficient cell lines. Eur J Immunol 2005; 35: 1250–6
- Li L, Moshous D, Zhou Y, Wang J, Xie G, Satido E, et al. A founder mutation in Artemis, a SNM1-like protein, causes SCID in Athabascan-speaking Native Americans. J Immunol 2002; 168: 6323–9
- Rooney S, Sekiguchi J, Zhu C, Cheng HL, Manis J, Whitlow S, et al. Leaky Scid phenotype associated with defective V(D)J coding end processing in Artemis-deficient mice. Mol Cell 2002; 10: 1379–90
- Moshous D, Pannetier C, Chasseval RR, Deilst FF, Cavazzana-Calvo M, Romana S, et al. Partial T and B lymphocyte immunodeficiency and predisposition to lymphoma in patients with hypomorphic mutations in Artemis. J Clin Invest 2003; 111: 381–7
- Ege M, Ma Y, Manfras B, Kalwak K, Lu H, Lieber MR, et al. Omenn syndrome due to ARTEMIS mutations. Blood 2005; 105: 4179–86
- Iwakawa M, Imai T, Harada Y, Ban S, Michikawa Y-I, Saegusa K, et al. Nippon Acta Radiol 2002; 62: 484–9
- West CM, McKay MJ, Holscher T, Baumann M, Stratford IJ, Bristow RG, et al. Molecular markers predicting radiotherapy response. Report and recommendations from an International Atomic Energy Agency technical meeting. Int J Radiat Oncol Biol Phys 2005; 62: 1264–73
- Baumann M, Holscher T, Begg AC. Towards genetic prediction of radiation responses ESTRO's GENEPI project. Radiother Oncol 2003; 69: 121–5
- Ho AY, Atencio DP, Peters S, Stock RG, Formenti SC, Cesaretti JA, et al. Genetic predictors of adverse radiotherapy effects: The Gene-PARE project. Int J Radiat Oncol Biol Phys 2006; 65: 646–53