Abstract
Introduction. Recent studies have demonstrated the capacity of the human organism to prevent the growth of potentially carcinogenic cells by paralyzing them. This antitumor mechanism is known as cellular senescence and is defined as an emergency defence system for cells on the way to becoming cancerous. Results. This review of the literature suggests that oncogene-induced senescence may be a response to oncogenic activation, acting as a natural barrier against tumorigenesis at a premalignant stage. Thus, a large number of cells enter senescence in premalignant lesions but none do so in malignant tumors, due to the loss of senescent pathway effectors such as p16INK4a or ARF-p53. Potential senescence markers in oral precancerous lesions include p21WAF1, p16INK4a, pRb, Maspin, RAR-β, G-actin, p15INK4b, DCR2, and DEC1, some of which are currently under study. Conclusion. In the short term, the study of this mechanism may yield valuable data for the management of oral cancer and precancer, for which no effective diagnostic or prognostic markers are yet available.
In developed countries head and neck cancer represents 5–10% of all malignant diseases. Over the past few decades, there has been a significant increase in oral cancer and related mortality in Europe, especially among young adults Citation[1]. In some developing countries, nearly half of cancer patients have oral cancer, largely due to exposure to carcinogens such as tobacco Citation[2]. In Spain, oral cancer was responsible for around 4% of cancer deaths in 2002, causing 1 884 deaths in males and 418 in females Citation[3]. In the European Union, 42 109 cases of oral cancer were recorded in males in 1998, with 15 744 deaths, and 11 447 cases in females, with 4 434 deaths Citation[4].
Most malignant tumors of the oral cavity are oral squamous cell carcinomas (OSCC). Over 300 000 new cases of OSCC are recorded annually each year worldwide. This aggressive epithelial neoplasm is associated with severe morbidity and a long-term survival of less than 50%, despite advances in surgical treatments in combination with radiotherapy and/or chemotherapy. Treatment outcomes would be improved if therapy could be applied before advanced stages are reached Citation[2]. Malignant disease is sometimes preceded by potentially malignant lesions, e.g., leukoplakia or erythroplakia. According to well-documented epidemiological data across different countries over the past 30 years, the prevalence of oral leukoplakia ranges from 1.1 to 11.7%, with a mean prevalence of 2.9%. In smoking patients, leukoplakia prevalence ranges from 3.7 to 60.3% Citation[5]. Up to 10% of patients with leukoplakia have an invasive carcinoma in the lesion Citation[6]. Prevention of leukoplakia malignization is essential, given the low survival of patients with oral cancers on leukoplakias Citation[7].
Oral carcinogenesis is widely accepted to be a highly complex molecular process comprising multiple steps in which genetic damage is accumulated, involving alterations of cells and of cytoplasmic signals that affect the cell cycle and DNA repair Citation[8]. Various molecular studies have reported that 6–10 genetic alterations are required for the malignant transformation of oral mucosal epithelial cells Citation[9].
Mutations are the main risk of genomic damage in mitotic cells. The genome is constantly affected by the environment and by-products of oxidation metabolism and, in the case of dividing cells, by errors in DNA replication and mitosis. Cells may attempt to repair damage or die, depending on its type and amount. Cancer development conditions are established when alterations confer advantages for growth and survival or when they make the genome unstable and therefore hypermutable.
Multicellular organisms have developed at least two cell mechanisms to impede the proliferation of cells at risk of oncogenic transformation, i.e., apoptosis (programmed cell death] or cellular senescence. Although these two mechanisms share characteristics in common, apoptosis kills and eliminates potentially cancerous cells, whereas cellular senescence irreversibly prevents their growth, presenting barriers that cells must overcome if they are to progress towards malignity.
The present review addresses the issue of cellular senescence in cancer and precancer and explores its possible connections with OSCC and premalignant lesions of the oral cavity.
Cellular, replicative and oncogene induced senescence
Many cells stop dividing not only when they are finally specialized and differentiated but also if they are under stress or receive some damage to their DNA. This permanent arrest of their replication and proliferation is known as replicative cellular senescence. On other occasions, cells that are damaged or no longer necessary undergo a process of apoptosis or programmed cell death, producing a characteristic phenotype. Normal cells, especially in humans, do not often undergo apoptosis in response to moderate DNA damage but rather respond by adopting a senescent phenotype Citation[10].
Cellular or replicative senescence (RS) was observed and proposed as a model of aging over 40 years ago by Hayflick and Moorhead Citation[11] in their study of fibroblasts obtained from in vitro skin and lung cultures. They observed that the cells divided but then stopped doing so as the culture aged. Besides this loss in dividing capacity, there was also a change in cell morphology. The authors established that cultures ceased dividing after a mean of 50 divisions, known as the Hayflick limit, Phase III phenomenon, or RS.
Most somatic cells in mammals, with the exception of germ and early embryonic cells, do not express telomerase (ribonucleoprotein complex that adds de novo telomeric repeats to chromosomes). Because DNA replication is bidirectional and DNA polymerases are unidirectional and require a “primer” or replication initiator (short and unstable RNA fragment), 50–200 pairs of telomeric DNA bases remain unreplicated at the end of the S phase. Hence, in absence of telomerase, telomeres are shortened at each cell division. When they reach a critical length, normal cells cease proliferation and acquire different morphological and functional characteristics in the RS response Citation[12].
There is considerable evidence that the senescence response evolved to suppress tumorigenesis, acting as a safety mechanism to prevent proliferation of cells at risk of neoplastic transformation Citation[13]. Accordingly, normal cells suffer senescent arrest when they receive stimuli capable of inducing or promoting neoplastic transformations. These stimuli include telomeres with compromised function, some types and levels of damage to DNA, perturbation of chromatin structure, and certain mutagenic transducer signals from oncogenes, e.g., mutated RAS. In fact, telomerase is incapable of preventing senescence in response to mutated RAS in human fibroblasts, indicating that cells can express a senescent phenotype without having functional telomeres, a phenomenon designated “stress-induced premature senescence (SIPS)”, “premature senescence”, or “accelerated senescence” Citation[14]. This contrasts with RS, which results from the physiological reduction of telomeres Citation[15–17]. The telomere shortening rate would itself be strongly influenced by oxidative cell stress Citation[14].
Senescent cells are resistant to apoptosis and have persisted in senescence in lab wacultures for up to 3 years. These cells are much more active metabolically than normal cells but do not divide. The senescent state cannot be reversed by physiological signals. Senescent cells have shown gene expressions not observed in young cells, although it is not known whether the expression precedes or follows the state of senescence. The new expression of genes in senescence represses growth factor (GF) transcription factors Citation[18]. Besides this repression of growth inducers, there is an activation of cell cycle inhibitors p21WAF1 and p16INK4a, genes that act to induce cell senescence and that are the final product of genetic programs leading cells into this state Citation[19].
Tumor cells are exposed to different stress situations, therefore senescence induction may represent an important arrest of tumor progression. One source of stress derives from aberrant proliferative signals from oncogenes, which can activate senescence via a process known as oncogene-induced senescence (OIS). This response may be critical for cell protection against cancer. However, investigation to date has been limited to cells in culture or in vivo studies of mice manipulated to over-express an oncogene (e.g., ras) Citation[20].
Numerous recent studies observed OIS during the earliest stages of tumor development in both animals and humans Citation[20], Citation[21], demonstrating that OIS restricts the growth of oncogenically stressed cells and thereby maintains the tumor in a premalignant, non-aggressive state. Conversely, absence of OIS due to the mutation of senescence induction pathways leaves the way open for oncogene-mediated malignant progression Citation[20–22]. The association between senescence and premalignant lesions (characterized by normal cell morphology and absence of invasive growth) makes senescence detection a promising biomarker that may serve as a prognostic indicator. However, it should be borne in mind that the presence of senescence in premalignant lesions is not incompatible with tumor growth, which depends on the balance between proliferation and apoptosis or senescence Citation[21].
A further question that has not yet been answered is how much oncogenic activity is required, not only to induce the senescence phenotype (OIS) but also to produce the malignant transformation of premalignant or normal tissue. Collado and Serrano proposed that oncogenic stress progressively increases during tumor development and that senescence is activated after the onset of tumors but before they acquire a malignant phenotype Citation[21].
Antagonistic pleiotropism
It has been proposed that senescence contributes to aging and to the onset of some diseases related to old-age Citation[12]. It is known that the senescent response in aging also results in changes to cell morphology and functionality. Due to senescence, some cell types resist certain apoptotic signals, which may explain why senescent cells accumulate in tissue with older age Citation[23]. Moreover, senescent cells tend to over-express secretion molecules, which can act at distant sites in tissue and affect the local microenvironment. These molecules include various matrix metalloproteinases and other degradative enzymes, inflammatory cytokines, and some GFs that establish the senescent phenotype of the cell Citation[24].
It may seem paradoxical that this process is simultaneously beneficial (preventing tumorogenesis) and detrimental (contributing to aging). Thus, cellular senescence appears to be an example of antagonistic pleiotropism. According to this theory, events selected by evolution to optimize the health of young adult organisms can also exert poorly selected harmful effects on the aged organism Citation[25]. These damaging effects are presumably negligible in young tissue, where senescent cells are rare. However, as the organism ages, senescent cells accumulate and there may be a change in their function, especially in their secretory phenotypes, which may compromise the physiology and integrity of the tissue Citation[10], Citation[26].
Mutations also accumulate over time, and there is an increasing probability with higher age that senescent cells and cells with oncogenic mutations will appear at around the same time. Consequently, senescent cells may create a microenvironment that promotes the growth and neoplastic progression of mutated cells Citation[27], Citation[28]. Thus, senescent cell cultures have shown expression of genes with different paracrine activities that produce growth factors and extracellular matrix or membrane receptor proteins. These include: Cyr61 protein, with mitogenic and angiogenic functions; prosaposin protein, with mitogenic and antiapoptotic functions; and TGFα and various proteases, which have the potential to facilitate metastatic growth Citation[29].
A major concern is that some chemotherapeutic agents that cause DNA damage also stimulate senescent cell development, which may have harmful local and systemic effects. There is an evident need to minimize or eliminate the adverse effects of senescence. However, it must first be more clearly understood why some cells follow the apoptosis pathway and others the senescence pathway and how the senescent phenotype and senescent cell-secreted factors are regulated Citation[30].
Cellular senescence pathways
Although various stimuli may trigger a senescence response, they all appear to converge in one or both of two pathways that establish and sustain senescent growth arrest. These pathways are regulated by p53 and retinoblastoma protein (pRB) tumor suppressor proteins Citation[26], Citation[30]. How these pathways are activated by the very different stimuli, whether both must be activated to trigger the senescence response, and how the senescence state is sustained remain unanswered questions.
ARF-p53 pathway
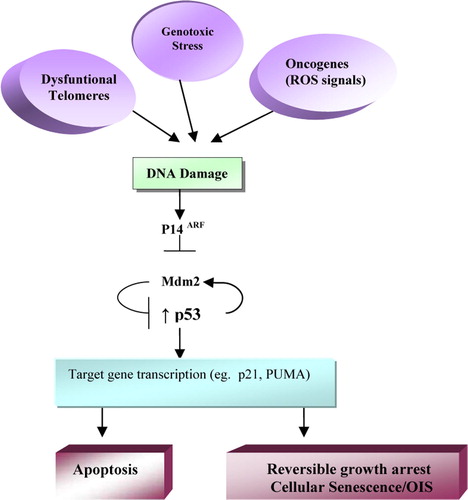
P53 is a crucial mediator of the cell response to DNA damage, including the senescence response, and loss of p53 function delays RS in some human cells, such as diploid fibroblasts Citation[10]. Dysfunctional telomeres also activate many components of the p53-mediated response, and the senescence response to dysfunctional telomeres requires the integrity of this pathway. Reactive oxygen species (ROS), which include oxygen ions and free radicals, have mutagenic effects on the RAS pathway and also induce cellular senescence Citation[31]. Thus, overexpressed oncogenic RAS may trigger a p53-dependent response, in this case due to the production by ROS of high DNA damage levels. However, oncogenic RAS can also induce p16INK4a, an activator of the pRB pathway, establishing a second barrier against the proliferation of potentially oncogenic cells. In some replicatively senescent human cells, such as BJ fibroblasts, p53 inactivation completely reverses senescent arrest Citation[32]. Likewise, cells enter telomere-dependent RS after inactivation of the gene that encodes for p21WAF1, a target for p53 transactivation and inhibitor of cell cycle progression. Therefore, in human fibroblasts, induction of senescence due to DNA damage, telomere dysfunction, and possibly oncogene overexpression converges in all of these cases in the ARF-p53 pathway, which is necessary and sufficient to establish and sustain senescence arrest (). Although this state of senescence cannot be reversed in these cells by physiological signals, it can be reversed by loss of p53 function Citation[13], Citation[30].
Figure 1. ARF-p53 senescence pathway. The p53-mediated response is activated by DNA damage, dysfunctional telomeres, and genotoxic stress. E.g., the ROS (Reactive Oxygen Species) produced by mitogenic signaling pathways. Proteín p14ARF inhibits mdm2 protein, which in turn favors degradation of p53. Transcription of genes that depend on p53, including the gene encoding for p21, induces senescent-type arrest of cell growth. This arrest cannot be reversed by physiological mitogens but is reversible by inactivation of p53. (Modified from Campisi, 2005)
P16INK4a-pRB pathway
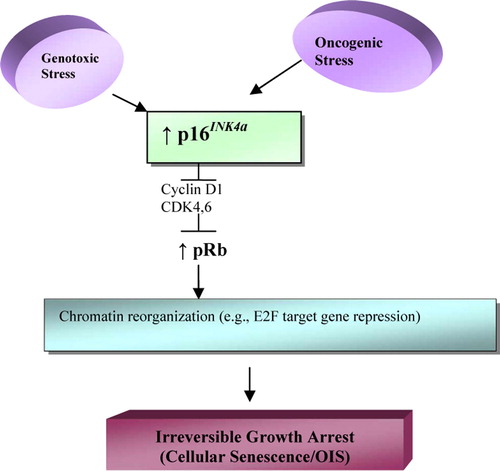
p53 inactivation reverses cell arrest in some cells but not in others. It has been observed that the latter express p16INK4a, cell cycle inhibitor and positive regulator of pRB. p16INK4a is induced by a wide variety of stressing stimuli, including overexpression of an oncogene, e.g., RAS. Cell cultures indicate that p16INK4a prevents senescence reversibility by p53-inactivation. Therefore, this tumor suppressant (p16INK4a) and, presumably, the pRB pathway it activates represent formidable barriers against cell proliferation that cannot be overcome by loss of p53 function Citation[30].
Cells entering senescence by the p16INK4a-pRb pathway remain in this (irreversible) state even when both p53 and pRb are inactivated Citation[30]. In the senescence process, pRB-E2F complexes, e.g., active pRb, produce the development of dense heterochromatin foci that repress various genes involved in cell cycle progression (). Moreover, once these heterochromatin foci are formed, they do not appear to require p16 INK4a or pRb activation to persist and can therefore permanently maintain the senescence state Citation[30].
Figure 2. p16INK4a-pRb senescence pathway. Oncogenes and other types of stress induce p16INK4a, activating pRb, which establishes repressive heterochromatin in E2F loci and possibly in other growth promoting genes. Once pRb mediated senescent arrest is established, it is not reversible by inactivation of p53 or pRb or both. (Modified from Campisi, 2005)
Senescence in oral cancer and precancer
Although oral leukoplakia has been considered a premalignant lesion for some decades, there are still no accurate prognostic and clinical indicators available for the reliable identification of potentially malignant lesions. Some clinical and histological characteristics have been related to the progression of oral leukoplakia lesions, but no correlation has been found between the different degrees of dysplasia and the prognosis. Thus, some mild dysplasias evolve into in situ carcinomas and some severe dysplasias do not Citation[2].
In fact, it has been reported that acquisition of the immortal phenotype is not related to histopathological status (i.e., degree of dysplasia), although this phenotype has been observed in early stages of oral carcinogenesis Citation[33]. Given the variability of these histopathological markers and the absence of correlations with clinical events, new diagnostic and prognostic molecular markers are necessary. From a clinical standpoint, genetic prognostic markers (DNA arrays) appear to be ideal future candidates. In the short- and medium-term, routine diagnoses are likely to be simplified by the development of molecular markers using immunohistochemistry (IHC).
It has also been proposed that senescent cells may stimulate cancer progression, which requires oncogenic mutations and an affected or damaged tissue environment in which the mutated cells can express their neoplastic phenotype Citation[27]. It was observed that senescent human fibroblasts stimulated the proliferation of preneoplastic but not normal epithelial cells in culture. Factors secreted by senescent cells were responsible for much of this stimulation Citation[27]. This situation is favored in advanced ages, with the accumulation of senescent cells and cells with preneoplastic mutations.
Senescent cells have been observed in precancerous lesions in lung, pancreas, and skin but not in carcinomas in the same organs Citation[20]. When a cell loses its senescence response, it appears to pass from a benign (premalignant lesion) to a malignant (carcinoma) state. However, given the complexity of the carcinogenic process, this should be regarded as another expression of carcinogenesis rather than as a cause-effect relationship Citation[20].
It has been experimentally established that the loss of senescent response in malignant lesions is correlated with absence of the main senescence effectors, e.g., p16 INK4a and p53. Conversely, using DNA microarrays, overregulation of a series of genes was observed in senescent cells in culture; the main proteins encoded were p15INK4b, Dec1, DcR2 Citation[20]. These markers were verified in premalignant lesions in skin, lung, and pancreas from mice with H-ras and K-ras mutations Citation[20], and may be also useful in oral premalignant lesions.
Oral carcinogenic progression markers and molecular pathways related to senescent pathways
As mentioned above, and due to the complexity of oral carcinogenesis, no definitive molecular markers are yet available to reliably differentiate among normal, premalignant, and malignant cells in the oral mucosa. An increase in the expression of some molecular markers (e.g. p53) has been observed with the progression of carcinogenesis, but there is no marker detected in precancerous lesions that does not also appear in malignant lesions. In a meta-analysis of seven studies, Warnakulasuriya et al. reported that p53 expression was over-expressed in 47% of premalignant oral lesions, much higher than the percentage of malignant transformation observed in premalignant lesions such as leukoplasia Citation[34].
Some studies have used IHC to demonstrate absence of mutated p53 protein in malignant head and neck lesions. However, the absence of mutated expression does not imply that p53 is normally expressed. The use of p21WAF1 as a prognostic marker has shown controversial results, with some authors describing its expression as favorable for the prognosis in head and neck cancer but others relating its overexpression to a more malignant tumor phenotype. Increases in the expression of EGFR and TGF-α have also been tested as progression markers, but the results obtained were unable to offer a clear identification of the state of malignization Citation[35].
Kang and Park established an in vitro model of oral carcinogenesis to describe the mechanisms by which environmental factors facilitate the appearance of oral cancer, stimulating normal human oral keratinocytes (HOKs) with high-risk human papillomavirus (HPV) and chemical carcinogens. After introduction of the HPV genome, cells bypassed the senescence checkpoint and entered into a prolonged but not immortal life cycle, during which telomeres continued to shorten. In a few immortal clones, they observed a marked elevation of telomerase activity (human telomerase reverse transcriptase [hTERT)]) and stabilization of telomere length. In addition, E6 and E7 oncoproteins of high risk HPV disrupted cell cycle control and DNA repair in immortalized HOKs, favoring mutations due to genomic instability, although this effect was only obtained when these proteins were introduced in combination with chemical carcinogens. They concluded that oral carcinogenesis is a series of discrete gene changes resulting from a continued genotoxic challenge by environmental risk factors Citation[36].
An immunohistochemical analysis of cyclin D1, p16INK4, and pRb in paraffin-embedded sections of 220 OSCCs, 90 leukoplasias, and 81 normal oral tissue samples found that the transition from a premalignant to malignant state was associated with the pRb-/cyclin D1+ phenotype (OR = 2.294, p = 0.001) and p53+ phenotype (OR = 2.230, p = 0.002). Multivariate analysis revealed that the pRb-/p53+ phenotype was related to a worse disease-free survival prognosis Citation[37].
summarizes the senescence markers most frequently implicated in senescence pathways Citation[20], Citation[28], Citation[30], Citation[38]. The pathway/phenotype markers that have demonstrated some relationship with the progression or inhibition of oral carcinogenesis are described below.
Table I. Current senescence markers possibly related to oral cancer and precancer
SA ß-gal
This lysosomal hydrolase, senescence-associated β-galactosidase (SA ß-gal), is elevated in senescent cells as a result of lysosomal activity at suboptimal pH (pH 6), which is detectable in senescent cells due to an increase in lysosomal content Citation[39]. It is considered an in vitro and in vivo senescence marker Citation[21].
P16 INK4a-Rb and ARF-p53 pathways
Investigations of signaling pathways that lead to OIS showed that the p16INK4a-Rb (retinoblastoma) and ARF-p53 pathways are responsible for the proliferation arrest that characterizes senescence Citation[30]. These two pathways are considered crucial for tumor suppression and are usually mutated in tumors. An elevation in p16INK4a, ARF, and p53 levels are related to OIS in animal tissue and human premalignant lesions. However, because this increased expression occurs at higher levels of the cascade (see ), there are situations in which tumor cells may have acquired mutations at lower levels that block senescence despite high p16INK4a and ARF-p53 expression Citation[40]. Thus, elevated levels of p16INK4a have been observed in dysplastic oral mucosa and in situ carcinomas but not in OSCC Citation[41]. On the contrary p16INK4a has also been described to be overexpressed in many head and neck cancers that contain HPV infection (6, 11,16 and 18 types) by tissue microarray Citation[42]. Elevated expression of mutated p53 protein has also been experimentally observed in dysplastic epithelial tissues, indicating that it is an early event in oral carcinogenesis Citation[43]. A study on head and neck tumors showed that the prevalence of p53 mutations before invasion did not increase as a function of the clinical stage of the tumor and that these changes therefore had little prognostic value Citation[44].
It is therefore important to find markers that, unlike p16INK4a and ARF-p53, are involved towards the end rather than the beginning of these senescence pathways Citation[21].
Senescence-associated heterochromatin foci (SAHFs)
Epigenetic changes associated with a global alteration in heterochromatin during senescence were recently related to the irreversibility that characterizes the senescence response Citation[20], Citation[45]. This alteration is initiated by pRb and results in a stable and permanent repression of genes that are crucial for proliferation, e.g., transcription factors of the E2F family. These genomic changes can be observed under the microscope and are known as SAHFs. Thus, the morphological appearance of senescent cell nuclei has been successfully used to identify the presence of OIS in vivo Citation[20], Citation[22].
Maspin
It was recently verified that maspin interacts with type I and III collagen, which is related to its antiangiogenic action in the extracellular matrix Citation[46]. Maspin expression does not appear to be universal. It is detected in specific cell types, e.g., myoepithelial cells in breast, basal cells in prostate, glandular epithelium and mucous cells in gastrointestinal tract, and epidermal layers in skin Citation[28]. Expression of this protein was found to gradually increase in normal human skin samples with higher age Citation[28]. However, it is not expressed in senescent fibroblasts, therefore it may be a marker that is mainly associated with epithelial tissue.
Telomerase
Telomerase is a ribonucleoprotein with enzymatic activity at nuclear level. It comprises two components: telomerase RNA (TR) and hTERT. Telomerase adds hexameric repeats of DNA (TTAGGG) to telomere terminals, compensating for the progressive loss of telomeric sequences inherent to DNA replication Citation[47]. Telomerase activity has been detected in various somatic tissues, including hematopoietic cells, basal keratinocytes, and basal endometrial, esophageal, and prostatic cells Citation[48]. Kang et al. reported that the RS of NHOKs is associated with a loss of telomerase activity followed by a limited telomere shortening Citation[49].
RAR-β
Retinoic Acid Receptor-β (RAR-β) has been used because retinoids induce the overregulation of some genes that induce senescence in breast cancer cells without causing side-effects (as opposed to p21WAF1-induced senescence) Citation[29]. RAR-β is dependent on retinoic acid in normal oral mucosa and is constitutively expressed in mortal dysplasia and completely suppressed in immortal dysplasia and oral carcinoma Citation[33], indicating the importance of RAR-β expression to maintain the mortal phenotype. The way in which retinoid receptors are implicated in senescence has not been elucidated, and their target genes have not been identified. It appears unlikely that p16INK4a is a target gene for RAR-β, because it is expressed in normal oral mucosa in absence of RAR-β Citation[33]. Higher RAR-β expression is observed when the cell approaches the final stages before senescence, whereas p16INK4a expression is high even in normal mucosa and is considered to play a major role in the initiation of senescence Citation[33].
p21WAF1
This protein is regulated (not exclusively) by P53. It inhibits cyclin/cyclin-dependent kinase (CDK) complex activities and induces senescent cells to express several genes that are detrimental for tumorigenesis control. Some of these encode for mitogenic and antiapoptotic proteins, such as prosaposin, TGFα, and βAPP (protein implicated in Alzheimer's disease), and other genes associated with aging-related diseases, e.g., amyloidosis, arteriosclerosis, and arthritis Citation[29]. One of the functions of p21WAF1 is to increase transcription factor NFκB by activating transcription of cofactors for histone deacetylase, p300, and CBP/a Citation[50].
PRb
Although the role of altered pRB expression in oral carcinogenesis has not been fully elucidated, there is abundant evidence that the pRB pathway is affected by alteration of either positive (e.g. Cyclin D1 and CDK6) or negative (e.g. p16INK4a and p21WAFI/Cip1/Sdi1) regulators. Thus, loss of pRb and accumulation of p53 (pRb-/p53 + ) are associated with the histological progression of tumors and the acquisition of invasive potential Citation[37].
G-actin
In vitro observations (cultures of senescent human fibroblasts) demonstrated that nuclear expression of G-actin is an even earlier marker of senescence than is endogenous growth of enzyme β-galactosidase enzyme (SA-β-gal) Citation[38].
By using DNA microarrays, it proved possible to select in vivo markers that show changes in gene expression that occur only during senescence and not when this senescence is inhibited. These markers have been tested in animal models of tumorigenesis produced by endogenous oncogenic activation of HRAS or KRAS. Among tested markers, p15INK4b, DCR2, and DEC-1 have shown increased expression in premalignant lesions of skin, lung, and pancreas but no or only a small increase in tumors Citation[20]. Therefore, they may also be useful markers of senescence in premalignant and/or malignant lesions of the oral cavity.
p15INK4b
This CDK inhibitor has been correlated with the presence of other senescence markers, especially SA- ß -galactosidase and SAHFs. It has also been reported to be involved in the cell growth arrest imposed during Ras-induced senescence and to inhibit transformation by Ras, at least in cell cultures Citation[51].
DCR2
This gene is induced by p53 and is one of the sentinel receptors of the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). DCR2 suppresses TRAIL-induced apoptosis and appears to regulate chemosensitivity Citation[52].
DEC1
This transcription factor is involved in the control of circadian rhythms and has also been implicated in different signaling pathways Citation[20].
The main advantage of these markers (p15INK4b, DCR2, and DEC1) is that, unlike SA-ß-gal or SAHFs, they can be readily detected in paraffin-embedded tissue using conventional immunohistochemical techniques.
The clinical utilization of OIS markers may be valuable to detect cancer in early stages, and loss of these markers indicates tumor progression to malignancy. Collado and Serrano developed the concept of a senescence index. Theoretically, tumors with a high senescence index have a better prognosis while those with a low index are associated with aggressive lesions requiring immediate surgery Citation[21].
Implications of senescence for cancer therapy
Until recently, it was believed that transformed cells were not capable of entering senescence. However, it is now acknowledged that this state can be induced in neoplastic cells by genetic manipulations and epigenetic factors, including conventional anti-cancer drugs, radiation, and differentiation agents.
Although inactivation of p53, p21WAF1 and p16INK4a has been observed in most cancers, it has also been demonstrated that their inactivation does not completely remove the senescence response, implying the participation of other, unknown pathways in senescence Citation[53], Citation[54]. cDNA microarrays have been used to observe some overregulated genes with growth-inhibiting functions that appear independent of p21WAF1, p53, and p16INK4a, including BTG1 (tumor suppressor), BTG2, EPLIN, Maspin, IGFBP-6, MIC-1, and amphiregulin Citation[29].
There is a need to develop a senescence-inducing agent that avoids the side effects associated with p21WAF1 and p16INK4a proteins, e.g., induction of protumorigenic factors. In this respect, induction of senescence by retinoid receptors has been proposed as good therapeutic option, since retinoids overregulate some senescence-inducing genes in breast cancer cells with no side-effects. Receptors include EPLIN (ubiquitous intracellular suppressor), FAT10 (ubiquitous intracellular suppressor), IGFBP3 (antimitotic and proapoptotic agent), and βIG-H3 (cell adhesion inhibitor) Citation[55].
However, prospects for this therapeutic line appear limited, because these retinoid receptors are not expressed in tumor samples in vivo, although these senescence genes may be induced by pathways other than the retinoid one Citation[16]. Therapies that enhance p21WAF1 or p16INK4a expression might have repercussions on cells that have not yet been transformed and are therefore not affected by the protumorigenic products that these proteins induce.
Hence, persistence of senescent cells in the tumor can be considered a double-edged sword. On one hand, senescent cells do not divide, and they even produce GFs that inhibit tumor growth. On the other hand, they may produce factors with mitogenic, antiapoptotic, and angiogenic activities. These protumor factors appear to be determined to a large extent by the expression of p21WAFI/Cip1/SdI1 and p16INK4a. For this reason, established tumors with cells that express high GF-inhibitor levels and low p21WAF1 and p16INK4a levels are considered to have a more favorable prognosis Citation[16].
Demethylating agents
The demethylating agent 5-aza-2-deoxycytidine (Aza-C) has been used to study the re-expression of RAR-β and p16INK4a genes that present their methylated promoters in dysplasia and oral cancer. The drug achieves re-expression of these genes in some dysplasias but not in carcinomas. This indicates that RAR-β and p16INK4a genes are inactivated in immortal dysplasias by methylation of the promoter but are irreversibly silenced, later in the progression of the cancer, by other changes. Aza-C also suppresses hTERT in immortal dysplasias but not in carcinomas Citation[33].
Telomerase
The barrier to proliferation posed by telomere shortening has been proposed as a tumor suppressor mechanism. However, given the complexity of telomerase regulation and the stability of the telomere, the role of telomerase may not be so direct. Depending on the context, telomere shortening appears to either protect from or promote cancer. Furthermore, the activity of telomerase in normal human cells is more common than was first thought Citation[15]. Nevertheless, the fact that high telomerase levels are expressed by 90% of human tumors but not by normal cells endows this enzyme with potential value as a diagnostic marker or therapeutic target.
Myc genes are frequently dysregulated in human tumors, and Myc overexpression may produce reactivation of telomerase and stabilization of telomeres. However, this would allow permanent proliferation and has been described as a strategy by which incipient carcinogenic cells escape senescence Citation[47].
Therapy with telomerase inhibitors is only potentially useful in tumors that already have short telomeres, given that long telomeres require a physiological time to shorten and for telomerase to be activated. Moreover, this therapy would not be useful when telomeres are activated via pathways other than the telomerase pathway (alternative telomere lengthening). A further concern is that telomerase inhibition may decrease regenerative potential and increase genetic instability.
P53
Both p53 and p16 are usually inactivated in carcinogenic cells. It has been observed in vitro that senescence induced by the RAS oncogene is dependent on functional p53. One option for inducing senescence in tumor cells may be p53 reactivation by the use of small molecules that have been demonstrated to “normalize” mutated p53 activity Citation[56], Citation[57].
Other treatments
Moderate doses of doxorubicin induced the senescent phenotype in 11 of 14 cell lines derived from varying types of solid human tumor Citation[53]. Other researchers reported induction of the senescent phenotype in different tumor cell lines treated with cisplatin, hydroxyurea, doxorubicin, camptotecin, or bromodeoxyuridine Citation[16], Citation[54].
Poele et al. conducted the first clinical investigation correlating the senescence response with the chemotherapeutic treatment of cancer patients. The senescent phenotype was detected by SA-β-gal, p53, and p16INK4a staining. Out of the 36 tumors treated with chemotherapy (cyclophosphamide, doxorubicin, and 5- fluorouracil), 41% showed SA-β-gal enzymatic activity. Importantly, SA-β-gal staining was confined to tumor cells and the adjacent normal tissue was completely negative. This may suggest that chemotherapy-induced senescence is specific to tumor cells Citation[54].
Presence of senescent cells in the tumor and the relative abundance of proteins produced by senescent cells are important biological factors that may have significant prognostic implications for progression of the disease. More aggressive tumors might contain very few or no senescent cells or, alternatively, a substantial fraction of senescent cells that express CDK inhibitors (p16INK4a or p21WAF1) alongside senescence-related tumor promoter factors overregulated by the CDK inhibitors. Conversely, a more favorable prognosis could be expected for tumors with senescent cells that express high levels of secreted growth inhibitors but low levels of p21WAF1 and p16INK4a Citation[16].
An important research challenge in oral cancer is posed by the lack of a reliable diagnostic or prognostic marker of leukoplastic lesions that usually precede OSCCs. The multifactorial and multi-step process of oral carcinogenesis probably involves the functional alteration of cell cycle regulators in combination with the loss of cell apoptosis and senescence signaling pathways. Therefore, besides in vitro studies of cell cultures, there is also a need for clinical studies using DNA arrays, as already performed in other human cancers. Novel diagnostic and therapeutic approaches could result from a greater knowledge of the molecular alterations and a better understanding of the consequences of cell cycle deregulation in malignant oral keratinocytes.
Conclusions
Cellular senescence, as irreversible cell cycle arrest, represents a safety program to limit the proliferation capacity of cells exposed to endogenous and exogenous stress signals. This review of the literature suggests that oncogene-induced senescence may be a response to oncogenic activation, acting as a natural barrier against tumorigenesis at a premalignant stage. Thus, a large number of cells enter senescence in premalignant lesions but none do so in malignant tumors, due to the loss of senescent pathway effectors such as p16INK4a or ARF-p53. A better understanding of the stimuli that induce cellular senescence could lead to their future use in cancer prevention and treatment, as is already the case with cellular apoptotic mechanisms. In the short term, study of this mechanism may yield valuable data for the management of oral cancer, for which no effective diagnostic or prognostic markers are yet available.
Acknowledgements
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Oliver RJ, Dearing J, Hindle I. Oral cancer in young adults: Report of three cases and review of the literature. Br Dent J 2000; 188: 362–5
- Sudbo J, Reith A. The evolution of predictive oncology and molecular-based therapy for oral cancer prevention. Int J Cancer 2005; 115: 339–45
- Centro Nacional de Epidemiología. Mortalidad por Cáncer y otras causas en España. http://193.146.50.130/htdocs/cancer//mort2002.txt. 2002. Ref Type: Electronic Citation.
- European Comission Health Monitoring Program. Comprehensive Cancer Monitoring Program in Europe-EUCAN. htpp://www.dep.iarc.fr/eucan/eucan.htm. 1998. Ref Type: Electronic Citation.
- Banoczy J, Gintner Z, Dombi C. Tobacco use and oral leukoplakia. J Dent Educ 2001; 65: 322–7
- Femiano F, Scully C. DNA cytometry of oral leukoplakia and oral lichen planus. Med Oral Patol Oral Cir Bucal 2005; 10(Suppl 1)E9–E14
- Scully C, Porter S. ABC of oral health. Oral cancer. BMJ 2000; 321(7253)97–100
- Bettendorf O, Piffko J, Bankfalvi A. Prognostic and predictive factors in oral squamous cell cancer: Important tools for planning individual therapy?. Oral Oncol 2004; 40: 110–9
- Werkmeister R, Brandt B, Joos U. Aberrations of erbB-1 and erbB-2 oncogenes in non-dysplastic leukoplakias of the oral cavity. Br J Oral Maxillofac Surg 1999; 37: 477–80
- Campisi J, Kim SH, Lim CS, Rubio M. Cellular senescence, cancer and aging: The telomere connection. Exp Gerontol 2001; 36: 1619–37
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res 1961; 25: 585–621
- Kim Sh SH, Kaminker P, Campisi J. Telomeres, aging and cancer: In search of a happy ending. Oncogene 2002; 21: 503–11
- Campisi J. Cancer, aging and cellular senescence. In Vivo 2000; 14: 183–8
- von Zglinicki T, Saretzki G, Ladhoff J, d'Adda dF, Jackson SP. Human cell senescence as a DNA damage response. Mech Ageing Dev 2005;126:111–7.
- Mathon NF, Lloyd AC. Cell senescence and cancer. Nat Rev Cancer 2001; 1: 203–13
- Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res 2003; 63: 2705–15
- Wei S, Wei S, Sedivy JM. Expression of catalytically active telomerase does not prevent premature senescence caused by overexpression of oncogenic Ha-Ras in normal human fibroblasts. Cancer Res 1999; 59: 1539–43
- Dimri GP, Campisi J. Altered profile of transcription factor-binding activities in senescent human fibroblasts. Exp Cell Res 1994; 212: 132–40
- Smith JR, Pereira-Smith OM. Replicative senescence: Implications for in vivo aging and tumor suppression. Science 1996; 273(5271)63–7
- Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Tumour biology: Senescence in premalignant tumours. Nature 2005; 436(7051)642
- Collado M, Serrano M. The power and the promise of oncogene-induced senescence markers. Nat Rev Cancer 2006; 6: 472–6
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005; 436(7051)660–5
- Choi J, Shendrik I, Peacocke M, Peehl D, Buttyan R, Ikeguchi EF, et al. Expression of senescence-associated beta-galactosidase in enlarged prostates from men with benign prostatic hyperplasia. Urology 2000; 56: 160–6
- Jennings BJ, Ozanne SE, Hales CN. Nutrition, oxidative damage, telomere shortening, and cellular senescence: Individual or connected agents of aging?. Mol Genet Metab 2000; 71: 32–42
- Kirkwood TB, Austad SN. Why do we age?. Nature 2000; 408(6809)233–8
- Campisi J, Warner RH. Aging in mitotic and post-mitotic cells. The role of DNA damage and repair in cell aging, BA Gilchrest, VA Bohr, 2001; 1–16
- Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc Natl Acad Sci USA 2001; 98: 12072–7
- Nickoloff BJ, Lingen MW, Chang BD, Shen M, Swift M, Curry J, et al. Tumor suppressor maspin is up-regulated during keratinocyte senescence, exerting a paracrine antiangiogenic activity. Cancer Res 2004; 64: 2956–61
- Chang BD, Swift ME, Shen M, Fang J, Broude EV, Roninson IB. Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc Natl Acad Sci USA 2002; 99: 389–94
- Campisi J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005; 120: 513–22
- Lee AC, Fenster BE, Ito H, Takeda K, Bae NS, Hirai T, et al. Ras proteins induce senescence by altering the intracellular levels of reactive oxygen species. J Biol Chem 1999; 274: 7936–40
- Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, et al. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J 2003; 22: 4212–22
- McGregor F, Muntoni A, Fleming J, Brown J, Felix DH, MacDonald DG, et al. Molecular changes associated with oral dysplasia progression and acquisition of immortality: Potential for its reversal by 5-azacytidine. Cancer Res 2002; 62: 4757–66
- Warnakulasuriya KA, Tavassoli M, Johnson NW. Relationship of p53 overexpression to other cell cycle regulatory proteins in oral squamous cell carcinoma. J Oral Pathol Med 1998; 27: 376–81
- Srinivasan M, Jewell SD. Evaluation of TGF-alpha and EGFR expression in oral leukoplakia and oral submucous fibrosis by quantitative immunohistochemistry. Oncology 2001; 61: 284–92
- Kang MK, Park NH. Conversion of normal to malignant phenotype: Telomere shortening, telomerase activation, and genomic instability during immortalization of human oral keratinocytes. Crit Rev Oral Biol Med 2001; 12: 38–54
- Soni S, Kaur J, Kumar A, Chakravarti N, Mathur M, Bahadur S, et al. Alterations of rb pathway components are frequent events in patients with oral epithelial dysplasia and predict clinical outcome in patients with squamous cell carcinoma. Oncology 2005; 68: 314–25
- Kwak IH, Kim HS, Choi OR, Ryu MS, Lim IK. Nuclear accumulation of globular actin as a cellular senescence marker. Cancer Res 2004; 64: 572–80
- Kurz DJ, Decary S, Hong Y, Erusalimsky JD. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci 2000; 113(Pt 20)3613–22
- Masumoto N, Fujii T, Ishikawa M, Saito M, Iwata T, Fukuchi T, et al. P16 overexpression and human papillomavirus infection in small cell carcinoma of the uterine cervix. Hum Pathol 2003; 34: 778–83
- Natarajan E, Saeb M, Crum CP, Woo SB, McKee PH, Rheinwald JG. Co-expression of p16(INK4A] and laminin 5 gamma2 by microinvasive and superficial squamous cell carcinomas in vivo and by migrating wound and senescent keratinocytes in culture. Am J Pathol 2003; 163: 477–91
- Konig F, Krekeler G, Honig JF, Cordon-Cardo C, Fischer G, Korabiowska M. Relation between human papillomavirus positivity and p16 expression in head and neck carcinomas--a tissue microarray study. Anticancer Res 2007; 27(1A)283–8
- Okazaki Y, Tanaka Y, Tonogi M, Yamane G. Investigation of environmental factors for diagnosing malignant potential in oral epithelial dysplasia. Oral Oncol 2002; 38: 562–73
- Bosch FX, Ritter D, Enders C, Flechtenmacher C, Abel U, Dietz A, et al. Head and neck tumor sites differ in prevalence and spectrum of p53 alterations but these have limited prognostic value. Int J Cancer 2004; 111: 530–8
- Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003; 113: 703–16
- Blacque OE, Worrall DM. Evidence for a direct interaction between the tumor suppressor serpin, maspin, and types I and III collagen. J Biol Chem 2002; 277: 10783–8
- Cerni C. Telomeres, telomerase, and myc. An update. Mutat Res 2000; 462: 31–47
- Harle-Bachor C, Boukamp P. Telomerase activity in the regenerative basal layer of the epidermis inhuman skin and in immortal and carcinoma-derived skin keratinocytes. Proc Natl Acad Sci USA 1996; 93: 6476–81
- Kang MK, Kameta A, Shin KH, Baluda MA, Park NH. Senescence occurs with hTERT repression and limited telomere shortening in human oral keratinocytes cultured with feeder cells. J Cell Physiol 2004; 199: 364–70
- Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ. Regulation of NF-kappaB by cyclin-dependent kinases associated with the p300 coactivator. Science 1997; 275(5299)523–7
- Malumbres M, Perez DC, I, Hernandez MI, Jimenez M, Corral T, Pellicer A. Cellular response to oncogenic ras involves induction of the Cdk4 and Cdk6 inhibitor p15(INK4b]. Mol Cell Biol 2000;20:2915–25.
- Liu X, Yue P, Khuri FR, Sun SY. Decoy receptor 2 (DcR2) is a p53 target gene and regulates chemosensitivity. Cancer Res 2005; 65: 9169–75
- Chang BD, Xuan Y, Broude EV, Zhu H, Schott B, Fang J, et al. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene 1999; 18: 4808–18
- te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res 2002; 62: 1876–83
- Dokmanovic M, Chang BD, Fang J, Roninson IB. Retinoid-induced growth arrest of breast carcinoma cells involves co-activation of multiple growth-inhibitory genes. Cancer Biol Ther 2002; 1: 24–7
- Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer 2001; 1: 68–76
- Foster BA, Coffey HA, Morin MJ, Rastinejad F. Pharmacological rescue of mutant p53 conformation and function. Science 1999; 286(5449)2507–10
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005; 436(7051)725–30