Abstract
Introduction. Genomic copy number changes are involved in the multi-step process transforming normal retina in retinoblastoma after RB1 mutational events. Previous studies on retinoblastoma samples led to a multi-step model in which after two successive RB1 mutations, further genomic changes accompany malignancy: 1q32.1 gain is followed by 6p22 gain, that in turn is followed by 16q22 loss and 2p24.1 gain. Retinoma is a benign variant of retinoblastoma that was initially considered a tumor regression, but recent evidences suggest that it rather represents a pre-malignant lesion. Genetic studies on retinoma tissue have rarely been performed. Materials and methods. We investigated by Real-Time qPCR, copy number changes of candidate genes located within the 4 hot-spot regions (MDM4 at 1q32.1, MYCN at 2p24.1, E2F3 at 6p22 and CDH11 at 16q22) in retina, retinoma and retinoblastoma tissues from two different patients. Results. Our results demonstrated that some copy number changes thought to belong to early (MDM4 gain) or late stage (MYCN and E2F3 gain) of retinoblastoma are already present in retinoma at the same (for MDM4) or at lower (for MYCN and E2F3) copy number variation respect to retinoblastoma. CDH11 copy number is not altered in the two retinoma samples, but gain is present in one of the two retinoblastomas. Discussion. Our results suggest that MDM4 gain may be involved in the early transition from normal retina to retinoma, while MYCN and E2F3 progressive gain may represent driving factors of tumor progression. These results also confirm the pre-malignant nature of retinoma.
Retinoblastoma (RB) is the most common pediatric intraocular neoplasm initiated by inactivation of both alleles of the RB1 tumor suppressor gene after two successive mutations (M1 and M2) Citation[1]. Although the loss of RB1 is a prerequisite for retinoblastoma initiation, further genomic changes (M3-Mn) may drive to malignancy by activating oncogenes and inactivating tumor suppressor genes Citation[2].
Previous studies performed on RB tumor samples by conventional/microarray comparative genomic hybridization (CGH) or quantitative multiplex polymerase chain reaction (QM-PCR) reported recurrent genomic gains/amplifications at 1q32, 2p24, 6p22 and losses at 16q22 Citation[3–9]. The characterization of these genomic imbalances have led to identify candidate oncogenes and tumor suppressors: MDM4 and KIF14 at 1q32, MYCN and DDX1 at 2p24, E2F3 and DEK at 6p22, CDH11 and RBL2 at 16q Citation[10]. On the basis of the frequencies of these genomic changes, a multi-step model for RB progression has been proposed Citation[9]. In this model, loss of both RB1 alleles (M1-M2) is not sufficient to initiate tumor formation. The two most common frequent genomic changes are gains at 1q32 and 6p22 and they are therefore considered M3 and M4 molecular events necessary to drive malignant transformation. 16q22 loss and 2p24 gain/amplification are less frequent and they are reported as alternate M5 events accompanying tumor progression Citation[9].
Retinoma is considered a benign variant of retinoblastoma based on clinical and histopathological evidence Citation[11], Citation[12]. The distinctive clinical characteristics include a translucent, greyish retinal mass protruding into the vitreous, “cottage-cheese calcification” (75%) and retinal pigment epithelial migration and proliferation (60%) Citation[11]. Histological features include foci of photoreceptor differentiation (fleurettes), abundant fibrillar eosinophilic stroma, absence of mitotic activity, foci of calcification Citation[12]. Individuals carrying a constitutive RB1 mutation can manifest either retinoma or retinoblastoma or, more rarely, a combination of both in different eyes or in the same eye as two separate foci Citation[13]. The reported proportion of stable retinomas among carriers of a germline RB1 mutation, ranges from 1.8 to 10% Citation[11–15]. These lesions have been initially considered spontaneous tumor regression because they resemble retinoblastoma that have involuted after radiotherapy and chemotherapy Citation[16–19]. However, Gallie and colleagues pointed out that these lesions did not show convincing clinical evidences of tumor regression and proposed that they rather represent a stage in the pathway to retinoblastoma development Citation[15–20]. There are few reported cases of clinically diagnosed retinomas that have undergone malignant transformation Citation[14–21].
Studies aimed at clarifying retinoma/retinoblastoma relationship at molecular level are rarely performed. This is principally due to the fact that retinoma tissue is very difficult to obtain since patients are not treated and retinoma/retinoblastoma mixed tissues are rarely reported in enucleated eyes. In three such cases, the expression of the pro-apoptotic neurotrophin receptor p75 (p75NTR) has been evaluated Citation[21]. The authors found that p75NTR protein was expressed in both normal retina and retinoma tissue, while it was absent in retinoblastoma, leading to the suggestion that p75NTR loss might accompany malignancy. p75NTR is a member of tumor necrosis family receptors which binds to neurotrophins, and plays a key role in maintaining the balance between survival and death in the developing retina Citation[22–26]. Although p75NTR expression is lost, no gene copy number changes have been detected in retinoblastoma suggesting that it could be due to point mutations or epigenetic events Citation[9].
In the present work, we used Real-Time qPCR to profile genomic imbalances at the four retinoblastoma hot-spot regions (1q32.1, 2p24.1, 6p22 and 16q22) in two human eye samples with areas of retinoma adjacent to retinoblastoma.
Materials and methods
Pathology and Immunohistochemistry
Eyes enucleated for retinoblastoma were obtained from the archives of the Department of Human Pathology and Oncology of the University of Siena (Siena, Italy). After surgery, enucleated eyes were immersion-fixed in buffered formalin for 48h. After fixation, sampling, paraffin embedding and cut were performed according to the usual pathological methods. In addition to clinically diagnosed retinoma (Case 1), 30 different eyes enucleated for retinoblastoma (at least two different paraffin blocks for each case, at least four different sections for each block) were analyzed by hematoxylin-eosin (H&E) staining and by p75NTR immunostaining to identify areas of retinoma. Five-micron-thick sections were deparaffinized and rehydrated. Endogenous peroxidase was blocked with 3% H2O2 in a Tris-buffered saline solution for 15 min, and non-specific binding was blocked with normal goat serum for 5 min. Slides were immersed in Target Retrieval Solution (DAKO Cytomation, Glostrup, D) for antigen unmasking and heated 3 times for 5 minutes in a micro-wave oven at 750 W. Slides were incubated with the primary monoclonal antibody for p75NTR (Novocastra Laboratories, Newcastle-upon-Tyne, UK) diluted 1:50 for 1 hour and with the primary monoclonal antibody for Ki67 diluted 1:100 (Lab Vision, Suffolk, UK) for 1 hour. Secondary antibody (Lab Vision, Ultra Vision Large Volume Detection System, anti-polyvalent, HRP, Fremont, CA, USA) incubation was performed for 20 min. The binding reaction was detected using 3,3′-diaminobenzidine (DAB) (DAKO Corporation, Carpinteria, CA, USA). Slides were then lightly counterstained with Harry's hematoxylin. Both internal negative (sclera cells) and omitted antibody controls were used. Retina ganglion cells were used as internal positive control.
Laser capture microdissection and DNA extraction from tissue samples
Retinoblastoma, retinoma and retina tissues were identified on H&E-stained sections. Five-micron-thick sections were deparaffinized, rehydrated and stained with Mayer hematoxylin and yellow eosin, dehydrated with xylene. Slides were observed through an inverse microscope. Cells of different tissues were isolated by laser capture microdissection (Arcturus PixCell II, MWG-Biotech, Florence, Italy). Selected cells adhere to the film on the bottom of the cap (Arcturus, MWG-Biotech) and are immediately transferred into a standard microcentrifuge tube containing digestion buffer and proteinase K (20 µg/ml) (Qiagen, Hilden, Germany). DNA was extracted by the use of QIAmp® DNA Micro Kit according to the manufacturer's protocol (Qiagen, Hilden, Germany). The Hoechest dye binding assay was used on a DyNA Quant™ 200 Fluorometer (GE Healthcare) to determine the appropriate DNA concentration.
Whole genome amplification
Whole genome amplification was performed using the GenomePlex® Complete Whole Genome Amplification (WGA) kit (Sigma-Aldrich, UK) according to the manufacturer's protocol. Briefly, after DNA extraction from microdissected tissue cells, 100 ng of template DNA were incubated at 95°C for 4 min in 1x fragmentation buffer, and the sample was cooled on ice. The sample was further incubated with the Library Preparation Buffer and Library Stabilization solution at 95°C for 2 min and the sample was cooled on ice. One microliter of Library Preparation Enzyme was added and the mix incubated at 16°C for 20 min, 24°C for 20 min, 37°C for 20 min, and 75°C for 5 min. The resulting sample was amplified using WGA polymerase, after initial denaturation at 95°C for 3 min and for 14 cycles at 94°C for 15 s and at 65°C for 5 min. Amplification products were purified using GenElute™ PCR Clean-up kit (Sigma-Aldrich) according to the instructions of the suppliers. The appropriate DNA concentration was determined by a DyNA Quant™ 200 Fluorometer (GE Healthcare).
Real-Time quantitative PCR
Real-Time quantitative polymerase chain reaction (PCR) was performed to detect genomic copy number imbalances. We used a pre-designed set of primers and probes specific for real time PCR experiments provided by the Assay-by-Design service (Applied Biosystems, Foster City, CA, http://www.products.appliedbiosystems.com). Primers and probes were designed for MDM4, MYCN, E2F3 and CDH11 (). PCR was carried out using an ABI prism 7000 (Applied Biosystems) in a 96-well optical plate with a final reaction volume of 50 µl. PCR reactions were prepared from a single Mix consisting of: 2X TaqMan Universal PCR Master Mix, 20X gene Assay Mix, 20X RNAaseP Mix (internal reference) and HPLC pure water. A total of 100 ng of DNA was dispensed in each sample well for triplicate reactions. Thermal cycling conditions included a pre-run of 2 min at 50°C and 10 min at 95°C. Cycle conditions were 40 cycles at 95°C for 15 s and 60°C for 1 min according to the TaqMan Universal PCR Protocol (Applied Biosystems). Normal retina has been used for the purpose of calibration. The starting copy number of the unknown samples was determined using the comparative Ct method, as previously described Citation[27].
Table I. Primers and probes sequences for Real-Time qPCR.
Results
Clinical description
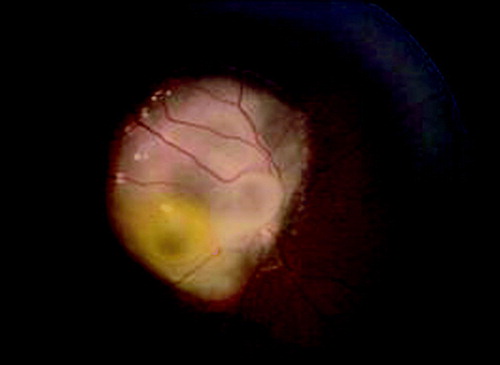
Case 1 is a 6 years and 3 months old male, third child of healthy and non-consanguineous parents. At the age of 1 year and 9 months strabismus was noted, but the parents referred that the first ophthalmoscopic evaluation was normal. At 2 years and 6 months, during periodical control, a retinoma was diagnosed in the right eye with the typical appearance of a whitish translucent mass without dilated vessels (). The retinal lesion remained stable for 11 months; the child underwent monthly examination under general anesthesia for the risk of malignant transformation of retinoma in early childhood. At 3 years and 5 months, during an ophthalmoscopic follow-up, a whitish creamy mass was noted on the retinoma surface with dilated feeder vessels indicating the malignant transformation. The retinoblastoma was firstly treated with conservative therapy, i.e. 4 cycles of chemoreduction (Carboplatin and Etoposide) combined with focal therapy (thermotherapy and argon laser therapy). A relapse was noticed 3 months later and other 2 cycles of chemotherapy were performed followed by stereotactic radiotherapy. The tumor remained in complete remission until the age of 5 years, when a new relapse appeared at the same site, so right eye enucleation was performed. During ophthalmoscopic follow-up no neoplastic foci were identified in the left eye.
Figure 1. Ophthalmoscopic examination of Case 1. Ophthalmoscopic examination at 2 years and 6 months showing cystic retinoma on the right fundus oculi overwhelming the optic nerve head.
Case 2 is a 3 years and 7 months old female, second child of healthy and non-consanguineous parents. At the age of 2 years and 5 months, her mother noted leukocoria in the right eye. At the ophthalmoscopic evaluation, a large tumor was present in the nasal area of the retina. Echografic examination showed the presence of a large mass with calcifications. Right eye enucleation was performed due to clinical diagnosis of eso- and endophytic retinoblastoma. The ophthalmoscopic follow-up did not identify any neoplastic focus in the remaining eye.
Histology
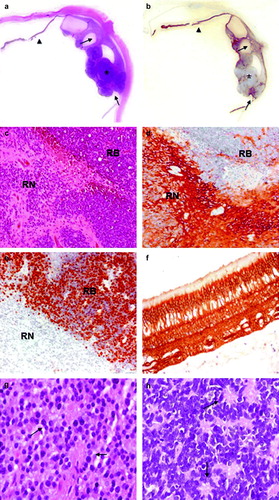
In Case 1 a relatively small, eso- and endophytic retinoblastoma was present in the posterior area of the eye infiltrating focally both the choroid and the prelaminar optic nerve (pT2c) (a). On the other side of the optic nerve, in part merged with RB cells, a cystic area was identified. Cells surrounding cystic spaces showed retinoma typical features, with plentiful cytoplasm, regular, smaller and light nuclei and signs of photoreceptor differentiation (fleurettes) (c and g). In contrast, RB tissue contained cells densely packed with little cytoplasm, partly arranged in Homer Wright rosettes (c and h).
Figure 2. Histology and immunohistochemistry of Case 1. a. H&E staining and b. p75NTR immunohistochemistry of the whole eye. Normal retina (arrow-head), retinoblastoma (asterisk) and retinoma (arrows) are shown. c. H&E staining (original magnification 100x) of retinoma (RN) and retinoblastoma (RB). d. Positive immunostaining for p75NTR (original magnification 100x) in retinoma (RN), while retinoblastoma cells (RB) are not stained as well as inflammatory cells inside the tumor. Spikes of positivity inside the tumor represent residues of neuron bodies and their elongation and vessel walls. e. Widespread Ki-67 nuclear cell immunostaining in RB cells indicates a high proliferation index, while retinoma cells are negative (original magnification 100X). f. Higher magnification of normal retina stained with p75NTR (original magnification 200X). p75NTR positivity in the retina is diffuse and includes neuron bodies and elongate processes of all the retina layers; outer segments of cones and rods are not stained. g. Higher magnification of retinoma (H&E staining, original magnification 200X) showing fleurettes (see arrows). h. Higher magnification of retinoblastoma (H&E stain, original magnification 200X) displaying Homer Wright rosettes (see arrows).
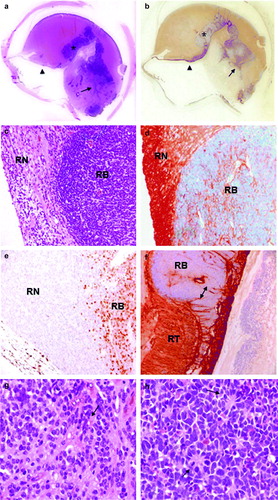
In Case 2 large, eso- and endophytic retinoblastoma was present in the posterior area of the eye (a). The tumor growth did not infiltrate the choroid nor the optic nerve (pT2a). In the central part of the neoplasm, in part merged with RB cells, a partly cystic area was identified. Cells surrounding cystic spaces showed abundant cytoplasm, regular, smaller and light nuclei, typical features of retinoma (c and g). Retinoma tissue also showed the presence of fleurettes, while RB cells were arranged in Homer Wright rosettes (g and h).
Figure 3. Histology and immunohistochemistry of Case 2. a. H&E staining and b. p75NTR immunohistochemistry of the whole eye. Normal retina (arrow-head), retinoblastoma (asterisk) and retinoma (arrow) are shown. c. H&E staining (original magnification 100x) of retinoma (RN) and retinoblastoma (RB). d. Positive immunostaining for p75NTR (original magnification 100x) in retinoma (RN), while retinoblastoma (RB) cells are not stained. Spikes of positivity inside the tumor represent residues of neuron bodies and their elongation and vessel walls. e. RB cells show a widespread Ki-67 nuclear cell immunostaining indicating a high proliferation index, while retinoma cells are not stained (original magnification 100X). f. Normal retina with an intraretinal RB focus stained with p75NTR (original magnification 200X). In the normal retina p75NTR positivity is diffuse, while in the neoplastic area positivity is restricted to vessel walls. g. Higher magnification of retinoma (H&E stain, original magnification 200X) showing fleurettes (see arrows). h. Higher magnification of retinoblastoma (H&E stain, original magnification 200X) with Homer Wright rosettes (see arrows).
p75NTR and Ki67 immunohistochemistry
In both cases, normal retina showed strong and consistent immunostaining positivity for p75NTR (b and f; b and f). The immunoreaction was located on the cellular surface of neuron bodies and their elongations. Differently, retinoblastoma cells showed absence of p75NTR staining (b and d; b and d). Immunopositivity was present in the vascular walls of veins, arteries and capillaries. In the areas occupied by RB cells, immunostaining was scanty and in the form of spikes of positivity, representing residues of neuron bodies and their elongations. Retinoma tissues adjacent to retinoblastoma were positive for p75NTR immunostaining (b and d; b and d). Furthermore, immunostaining for the proliferation marker Ki67, showed positivity in retinoblastomas and undetectable signal in retinomas of both cases (e and 3e). Such immunohistochemical features supported the morphological diagnosis of retinoma.
Gain and loss of chromosome regions
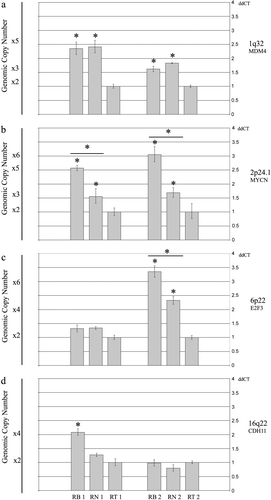
To identify genomic differences between retinoma and retinoblastoma, we profiled genomic copy number changes at four gain-loss “hot-spot” regions by Real Time qPCR in retina, retinoma and retinoblastoma tissues from two different patients. To avoid tissue contamination, we isolated retina, retinoma and retinoblastoma cells by laser capture microdissection. Due to the low amount of DNA obtained from microdissected paraffin-embedded tissues, we used whole genome amplification to pre-amplify DNA samples. Probes and primers for qPCR were designed on the following candidate oncogenes/oncosuppressors because literature data on their function or connection to p53 pathway inactivation were intriguing regard to a possible involvement in retinoma/RB transition: MDM4 (1q32.1), MYCN (2p24.1), E2F3 (6p22) and CDH11 (16q22) Citation[10–28]. In both cases, retinoblastoma and retinoma showed the same level of MDM4 gain respect to retina (for Case 1, ddCT ratio: 2.35 ±0.14 in RB and 2.41±0.13 in RN, indicating 5 copies; for Case 2, ddCT ratio: 1.62 ±0.09 in RB and 1.83±0.01 in RN, indicating 3 copies) (a). In the two cases, progressive MYCN copy number gain was detected in retinoma (3 copies) and in retinoblastoma (5 copies in Case 1 and 6 copies in Case 2) (b). ddCT ratio values were 2.57±0.05 (RB) and 1.55±0.24 (RN) in Case 1 and 3.05±0.13 (RB) and 1.69±0.14 (RN) in Case 2. For E2F3, in Case 1 no copy number changes were detected (ddCT ratio: 1.32±0.13 in RB and 1.34±0.06 in RN), while in Case 2, we identified copy number gain in retinoma (5 copies) and amplification in retinoblastoma (7 copies) (ddCT ratio: 3.36±0.09 in RB and 2.32±0.09 in RN) (c). For CDH11, copy number gain (4 copies) was detected only in retinoblastoma of Case 1 (d). ddCT ratio values were: 2.08 ±0.08 (RB) and 1.27±0.07 (RN) in Case 1 and 0.98±0.16 (RB) and 0.80±0.20 (RN) in Case 2.
Figure 4. Genomic changes detected by Real-Time quantitative PCR. ddCT ratios and standard deviations obtained for retinoma (RN), retinoblastoma (RB) and retina (RT) of Case 1 (left) and Case 2 (right). For each graph, genomic copy number is indicated on the left (gain: 3-7 copies; amplification ≥7 copies). The asterisk shows when the difference is statistically significant (t-test, p ≥ 0.05). a. Both cases show MDM4 gain in retinoma and retinoblastoma at the same number of copies. b. Progressive gain of MYCN from retinoma to retinoblastoma respect to normal retina in both cases. c. Case 1 does not show copy number changes, while Case 2 displays gain of E2F3 in retinoma and amplification in retinoblastoma compared to retina. d. No genomic changes were detected in the two retinoma tissues. One of the two retinoblastoma samples (Case 1) presents gain of CDH11.
Discussion
Retinoma is a retinal lesion highly associated with retinoblastoma but lacking malignant characteristics Citation[11]. These lesions have been initially called “spontaneous regression” of retinoblastoma. However clinical evidences do not support this hypothesis and suggest that retinoma rather represents a step towards retinoblastoma development Citation[11–21]. Very recently, during the revision process of the present manuscript, Dimaras and colleagues published interesting data clarifying this issue Citation[29]. They demonstrated that retinomas display inactivation of both RB1 alleles, absence of proliferative markers (Ki67 staining), low level of genomic instability and high expression of the senescence-associated proteins (p16INK4a and p130). Diversely, adjacent retinoblastomas show reduced expression of p16INK4a and p130 and increased genomic changes, indicating progression from retinoma.
Here, we investigated copy number changes at four “hot spot” regions for tumor development in two human eye samples with areas of retinoma adjacent to retinoblastoma. In the first case, there was clinical evidence of the presence of a cystic retinoma, already decribed by L. Zografos Citation[30], that later progressed to retinoblastoma. The second case of retinoma was identified by retrospective histopathological review of 30 eye samples enucleated for retinoblastoma (1/30, 3%). This percentage of retinomas in enucleated eyes is lower respect to the percentage reported by Dimaras (20/128, 15.6%), but this discrepancy could be due to the different number of reviewed eye samples Citation[29]. In both our cases, immunostaining for the pro-apoptotic neurotrophin receptor p75NTR and the proliferation marker Ki67 clearly distinguished retinoma areas from retinoblastoma, in accordance with previous data Citation[21–29].
Genomic copy number changes represent fundamental steps in the development of retinoblastoma Citation[9]. To detect such changes at the four “hot spot” loci, we employed Real Time quantitative PCR. This technique, differently from FISH, does not allow an analysis at single cell level but determines gene copy number on a whole tissue basis. Quantitative PCR allowed us to clearly detect the relative proportion of gene copy number changes between the three tissues: retina, retinoma and retinoblastoma. Our results showed progressive gain of MYCN (2p24) and E2F3 (6p22) copy number from retinoma to retinoblastoma, confirming that retinoma represents a transition step to tumor development (b and c).
Since both Case 1 and 2 display progressive MYCN gain from retinoma to retinoblastoma, these results emphasize the role of MYCN in malignant progression. In a previous study, Bowles et al. showed amplification of MYCN in 3% of 87 primary retinoblastoma and low-level gains in further 13% Citation[9]. They also found that MYCN gains/amplifications are more common in cell lines that in primary retinoblastoma suggesting that this copy number changes confer a selective advantage for cell growth Citation[9]. This characteristic could represent a driving force in retinoma/retinoblastoma transition.
Furthermore, in one of the two cases, we found E2F3 gain in retinoma and amplification in retinoblastoma. Gain of E2F3 gene has been found in 70% of retinoblastoma primary tumors Citation[9]. E2F3, together with E2F1 and E2F2, belongs to a subclass of E2F factors that act as transcriptional activators through the RB1-dependent G1/S phase transition Citation[31]. Moreover E2F3 represses p14/ARF, an important tumor repressor in the p53 pathway Citation[32], Citation[33]. Consequently E2F3 amplification may contribute to retinoblastoma growth from retinoma by eliminating p53 apoptotic process.
MDM4 showed gain in both retinoma and retinoblastoma of both cases, at the same number of copies. A recent study revealed an increased MDM4 copy number in 65% of human retinoblastoma Citation[28]. Our results not only confirm the importance of MDM4 gain in retinoblastoma development, but interestingly suggest that it may be involved in the early transition to retinoma (a). MDM4 is a negative regulator of p53 transcription and stabilizes the E3 ubiquitin ligase MDM2 which tags p53 for degradation Citation[34]. Increased p53 degradation could be one mechanism that contributes to inactivate the apoptotic process in retinoblastoma. Functional studies are necessary to confirm the involvement of MDM4 in the early phases of malignant progression and whether it represents a “driving” gene or a “passenger” gene whose amplification is a consequence of the amplification of another strong candidate located at 1q, KIF14. In fact, Dimaras et al. recently reported that KIF14 shows gains even more frequently and at higher levels respect to MDM4 Citation[29].
Since loss of CDH11 has been reported in 45% of primary retinoblastoma Citation[35], this gene has been proposed as candidate oncosuppressor. However, we did not find CDH11 loss. On the contrary, we identified copy number gain in one case of retinoblastoma (d). CDH11 overexpression has been reported in other tumor types like prostate cancer, rhabdomyosarcoma and invasive breast cancer Citation[36–38], suggesting that also the overexpression may contribute to cancerogenesis.
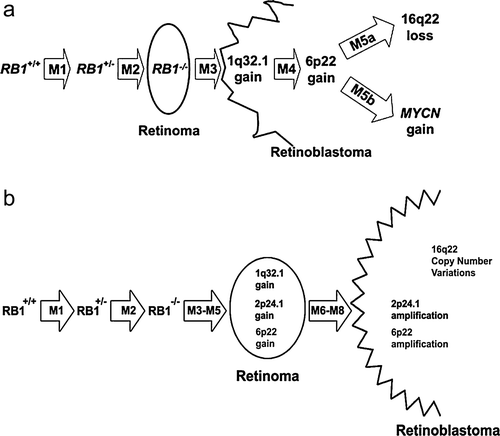
The results reported above significantly change the multi-step model of retinoblastoma progression proposed by Bowles on the basis of the frequency of genomic changes found in retinoblastoma tissues Citation[9]. According to this model, after two successive RB1 mutations (M1 and M2), the following further genomic changes accompany malignancy: 1q32.1 gain (M3), 6p22 gain (M4), 16q22 loss (M5a) and 2p24.1 gain (M5b) (a). In accordance with very rencent data, we found that 1q32.1 gains, thought to belong to early stages of retinoblastoma, are already present in retinoma Citation[29]. More interestingly, even the 6p22 and 2p24 gains thought to belong to later retinoblastoma stages are already present in retinoma, although at lower number of copies respect to retinoblastoma (b). In accordance with the previous model, no genomic changes were detected at 16q22 region in the two retinomas (). In one case, we found 16q22 gain in retinoblastoma tissue, confirming that copy number variations in this region are involved in later stages of tumor progression (d) Citation[9].
Figure 5. a. Tumor multi-step model from Bowles et al., 2007. b. Retinoblastoma tumor progression model based on our results. In light grey, copy number changes found in only one of the two samples.
It is important to note that, while Case 2 was enucleated at diagnosis, Case 1 was treated by chemotherapy and radiotherapy before enucleation and this may have at least in part influenced the pattern of genetic alterations. Further studies are therefore necessary to confirm these data. Unfortunately they are limited by sample availability. Only a collaborative effort by different centers will allow to perform these studies on a larger number of samples and to definitively characterize molecular events underlying retinoma and retinoblastoma. The accomplishment of this goal is important not only because it will clarify the pathogenic mechanisms of tumor development, but also because it will lead to the discovery of targets that will represent the basis of therapies aimed to control the malignant progression of retinoma.
Acknowledgements
This work was supported by a FIRB grant (RBIP00PMF2) to A.R., by the University of Siena grant PAR 2006 to M.B. and by a grant on Retinoblastoma from Istituto Toscano Tumori (ITT) to A.R. We thank Lucia Loiacono for technical support.
References
- Knudson AG, Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc Natl Acad Sci USA 1971; 68: 820–3
- Gallie BL, Campbell C, Devlin H, Duckett A, Squire JA. Developmental basis of retinal-specific induction of cancer by RB mutation. Cancer Res 1999; 59: 1731s–1735s
- Mairal A, Pinglier E, Gilbert E, Peter M, Validire P, Desjardins L, et al. Detection of chromosome imbalances in retinoblastoma by parallel karyotype and CGH analyses. Genes Chromosome Cancer 2000; 28: 370–9
- Chen D, Gallie BL, Squire JA. Minimal regions of chromosomal imbalance in retinoblastoma detected by comparative genomic hybridization. Cancer Genet Cytogenet 2001; 129: 57–63
- Herzog S, Lohmann DR, Buiting K, Schuler A, Horsthemke B, Rehder H, et al. Marked differences in unilateral isolated retinoblastomas from young and older children studied by comparative genomic hybridization. Hum Genet 2001; 108: 98–104
- Lillington DM, Kingston JE, Coen PG, Price E, Hungerford J, Domizio P, et al. Comparative genomic hybridization of 49 primary retinoblastoma tumors identifies chromosomal regions associated with histopathology, progression, and patient outcome. Genes Chromosome Cancer 2003; 36: 121–8
- van der Wal JE, Hermsen MA, Gille HJ, Schouten-Van Meeteren NY, Moll AC, Imhof SM, et al. Comparative genomic hybridisation divides retinoblastomas into a high and a low level chromosomal instability group. J Clin Pathol 2003; 56: 26–30
- Zielinski B, Gratias S, Toedt G, Mendrzyk F, Stange DE, Radlwimmer B, et al. Detection of chromosomal imbalances in retinoblastoma by matrix-based comparative genomic hybridization. Genes Chromosome Cancer 2005; 43: 294–301
- Bowles E, Corson TW, Bayani J, Squire JA, Wong N, Lai PB, et al. Profiling genomic copy number changes in retinoblastoma beyond loss of RB1. Genes Chromosome Cancer 2007; 46: 118–29
- Corson TW, Gallie BL. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosome Cancer 2007; 46: 617–34
- Gallie BL, Ellsworth RM, Abramson DH, Phillips RA. Retinoma: Spontaneous regression of retinoblastoma or benign manifestation of the mutation?. Br J Cancer 1982; 45: 513–21
- Margo C, Hidayat A, Kopelman J, Zimmerman LE. Retinocytoma. A benign variant of retinoblastoma. Arch Ophthalmol 1983; 101: 1519–31
- Sampieri K, Hadjistilianou T, Mari F, Speciale C, Mencarelli MA, Cetta F, et al. Mutational screening of the RB1 gene in Italian patients with retinoblastoma reveals 11 novel mutations. J Hum Genet 2006; 51: 209–16
- Balmer A, Munier F, Gailloud C. Retinoma. Case studies. Ophthalmic Paediatr Genet 1991; 12: 131–7
- Eagle RC, Jr, Shields JA, Donoso L, Milner RS. Malignant transformation of spontaneously regressed retinoblastoma, retinoma/retinocytoma variant. Ophthalmology 1989; 96: 1389–95
- Rubin ML. The tale of the warped cornea: A real-life melodrama. Arch Ophthalmol 1967; 77: 711–2
- Brockhurst RJ, Donaldson DD. Spontaneous resolution of probable retinoblastoma. Arch Ophthalmol 1970; 84: 388–9
- Morris WE, LaPiana FG. Spontaneous regression of bilateral multifocal retinoblastoma with preservation of normal visual acuity. Ann Ophthalmol 1974; 6: 1192–4
- Reese PD. The general ophthalmological examination for the non-ophthalmologist. J Ark Med Soc 1976; 72: 387–90
- Singh AD, Santos CM, Shields CL, Shields JA, Eagle RC, Jr. Observations on 17 patients with retinocytoma. Arch Ophthalmol 2000; 118: 199–205
- Dimaras H, Coburn B, Pajovic S, Gallie BL. Loss of p75 neurotrophin receptor expression accompanies malignant progression to human and murine retinoblastoma. Mol Carcinog 2006; 45: 333–43
- Rodriguez-Tebar A, Dechant G, Barde YA. Binding of brain-derived neurotrophic factor to the nerve growth factor receptor. Neuron 1990; 4: 487–92
- Ernfors P, Ibanez CF, Ebendal T, Olson L, Persson H. Molecular cloning and neurotrophic activities of a protein with structural similarities to nerve growth factor: developmental and topographical expression in the brain. Proc Natl Acad Sci USA 1990; 87: 5454–8
- Squinto SP, Stitt TN, Aldrich TH, Davis S, Bianco SM, Radziejewski C, et al. trkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3 but not nerve growth factor. Cell 1991; 65: 885–93
- Hallbook F, Ibanez CF, Persson H. Evolutionary studies of the nerve growth factor family reveal a novel member abundantly expressed in Xenopus ovary. Neuron 1991; 6: 845–58
- Frade JM, Barde YA. Genetic evidence for cell death mediated by nerve growth factor and the neurotrophin receptor p75 in the developing mouse retina and spinal cord. Development 1999; 126: 683–90
- Livak K. ABI Prism 7700 Sequence Detection System, 1997.
- Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, et al. Inactivation of the p53 pathway in retinoblastoma. Nature 2006; 444: 61–6
- Dimaras H, Khetan V, Halliday W, Orlic M, Prigoda NL, Piovesan B, et al Loss of RB1 induces non-proliferative retinoma; increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet 2008.
- Zografos L. Tumeurs intraoculaires. París: Société Française d'Ophtalmologie, Masson, 2002.
- Saavedra HI, Wu L, de Bruin A, Timmers C, Rosol TJ, Weinstein M, et al. Specificity of E2F1, E2F2, and E2F3 in mediating phenotypes induced by loss of Rb. Cell Growth Differ 2002; 13: 215–25
- Parisi T, Pollice A, Di Cristofano A, Calabro V, La Mantia G. Transcriptional regulation of the human tumor suppressor p14(ARF) by E2F1, E2F2, E2F3, and Sp1-like factors. Biochem Biophys Res Commun 2002; 291: 1138–45
- Ginsberg D. E2F3-a novel repressor of the ARF/p53 pathway. Dev Cell 2004; 6: 742–3
- Marine JC, Jochemsen AG. Mdmx and Mdm2: Brothers in arms?. Cell Cycle 2004; 3: 900–4
- Marchong MN, Chen D, Corson TW, Lee C, Harmandayan M, Bowles E, et al. Minimal 16q genomic loss implicates cadherin-11 in retinoblastoma. Mol Cancer Res 2004; 2: 495–503
- Tomita K, van Bokhoven A, van Leenders GJ, Ruijter ET, Jansen CF, Bussemakers MJ, et al. Cadherin switching in human prostate cancer progression. Cancer Res 2000; 60: 3650–4
- Markus MA, Reichmuth C, Atkinson MJ, Reich U, Hoffmann I, Balling R, et al. Cadherin- 11 is highly expressed in rhabdomyosarcomas and during differentiation of myoblasts in vitro. J Pathol 1999; 187: 164–72
- Pishvaian MJ, Feltes CM, Thompson P, Bussemakers MJ, Schalken JA, Byers SW. Cadherin-11 is expressed in invasive breast cancer cell lines. Cancer Res 1999; 59: 947–52