Abstract
Apoptosis is a physiological process vital for embryologic development and the maintenance of homeostasis in multicellular organisms, but it is also involved in a wide range of pathological processes, including cancer. In mammalian cells, apoptosis has been divided into two major pathways: the extrinsic pathway, activated by proapoptotic receptor signals at the cellular surface, and the intrinsic pathway, which involves the disruption of mitochondrial membrane integrity. Although many of the proteins vital for apoptosis have been identified, the molecular pathways of cellular death still remain to be elucidated. This review provides references concerning the apoptotic molecules, their interactions, the mechanisms involved in apoptosis resistance, and also the modulation of apoptosis for the treatment of cancer.
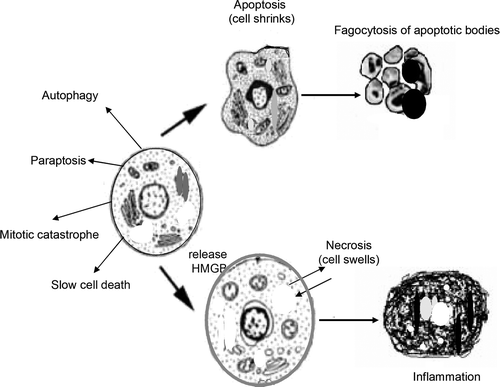
Programmed cell death (PCD) is a major component of normal development, the maintenance of homeostasis, and elimination of damaged cells in unicellular and multicellular organisms. Depending on morphological alterations and caspase involvement, cell death can be classified into different types: apoptosis, autophagy, necrosis, mitotic catastrophe, paraptosis, and slow cell death Citation[1].
Apoptosis, a controlled and energy-dependent process, is the best-described form of programmed cell death. Its deregulation can lead to cancer, autoimmune and degenerative diseases, explaining the increasing interest in elucidating the apoptosis pathways for disease etiology and therapeutic modulation. Many of the morphological changes that cells undergo during apoptosis have been observed by light and electron microscopy, including cell shrinkage and chromatin condensation with pyknosis formation. The most important characteristic features of apoptosis include cytoplasmic membrane blebbing, formation of apoptotic bodies and the phagocytosis of apoptotic bodies by neighboring cells or macrophages.
For a long time necrosis has been considered a passive and uncontrolled process of cell death. However, evidence is accumulating that necrosis may be as well-regulated as apoptosis Citation[2]. Necrosis often results in inflammation and adaptive immunity, whereas no inflammatory reaction is associated with apoptosis. One explanation for this difference in behavior is that apoptotic cells do not release their cellular constituents into the surrounding tissue and are quickly phagocytosed by macrophages and parenchymal cells without the production of inflammatory cytokines Citation[3]. Whether a cell dies by necrosis or apoptosis partially depends on the nature of the cell death signal, the tissue type, the developmental stage of the tissue and the physiologic milieu. More recently, a molecule called high mobility group box-1 protein (HMGB1), released by dying cells, has been shown to determine whether the immune system is activated or not Citation[4]. Necrotic cells, but not apoptotic cells, release HMGB1, which stimulates the immune system to produce inflammatory cytokines.
The immunosuppressive effect observed during apoptosis is not only mediated by soluble factors released from the dying cells. A new concept has been developed regarding the suppression of the response of phagocytes and non-classical phagocytes (including epithelial, endothelial, and fibroblastic cells) following engulfment of the apoptotic corpse Citation[5].
Autophagy, from the Greek words auto meaning self and phagein meaning eating (“self-eating”), is a lysosomal degradation pathway essential for homeostasis under normal conditions. In addition, autophagy can assume the cell-killing role when apoptosis is not an option Citation[6]. Autophagy is induced at low levels under normal conditions, but it becomes exacerbated in response to genotoxic or ER stress stimuli during apoptosis-deficient conditions Citation[7]. On the other hand, autophagy suppresses apoptosis in Myc-dependent lymphomas, inducing tumor growth and promoting the survival of cancer cells that express the apoptosis-inhibiting Bcl2 family under hypoxic conditions Citation[8], Citation[9]. These data suggest a complex role for autophagy in tumor formation and that its inhibition might provide therapeutic benefit ().
Figure 1. Various models of cell death, including apoptosis and necrosis, depend on HMGB protein release with the death stimulus. Other models have been proposed to define the caspase-independent pathway of cell death: paraptosis, autophagy, mitotic catastrophe and slow cell death.
Mechanisms of apoptosis
The two apoptosis pathways are the extrinsic and intrinsic pathways. The extrinsic pathway operates via death receptors on the cell surface, and the intrinsic pathway depending on mitochondria is activated by loss of growth factor signals or in response to lethal stimuli from inside the cell. Both pathways are interconnected with other signaling proteins, such as NK-κB and p53-MDM2, and converge at the level of the effectors proteolytic enzymes, called caspases.
The caspase cascade
The process of apoptosis is carefully controlled, involving an energy-dependent cascade of molecular events. The morphological changes of apoptosis are due to the action of well-preserved and efficient cysteinyl aspartate-specific proteases called caspases. Caspases are widely expressed in most cells in an inactive proenzyme form, and when activated by proteolytic processing they activate other procaspases in turn, thus amplifying the apoptotic signaling pathway and leading to cell death Citation[10]. So far, 14 caspases have been identified, 11 of which are present in mammals. They are subdivided, depending on their activity, into proinflammatory (caspases 1, 4, 5, 11, 12, 13 and 14) and proapoptotic caspases. The latter are grouped as initiator caspases (2, 8, 9, 10) and effector caspases (3, 6, 7). Little is known about some proinflammatory caspases, such as caspase 11, which has been reported to regulate apoptosis and cytokine maturation during septic shock Citation[11]; caspase-12, which mediates endoplasmic-specific apoptosis in mice and rats; caspase 13, which is found only in cattle; and caspase 14, which is highly expressed in embryonic tissues.
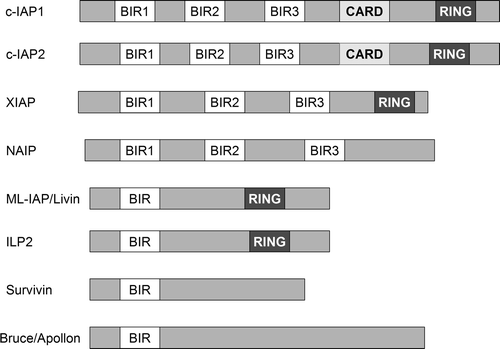
The caspases are modulated by several endogenous cellular factors, including the inhibitor of apoptosis (IAP) proteins. These polypeptides have a common amino-terminal, 70-residue domain, called the baculovirus inhibitor repeat (BIR) domain, which appears to bind and inhibit active caspases. There are eight mammalian BIR-containing proteins (BIRPs) identified thus far, including the inhibitors of apoptosis 1 and 2 (IAP1 and IAP2), X-linked inhibitor of apoptosis (XIAP), survivin, neuronal inhibitor of apoptosis (NIAP), and BIR-repeat-containing ubiquitin-conjugating enzyme (BRUCE) Citation[12]. Livin, also known as melanoma-specific inhibitor of apoptosis protein (ML-IAP), has been identified as a new member of the IAP family. Recent studies suggest that ML-IPA is expressed in some tumor cells and several fetal tissues and that it might regulate apoptosis by sequestering Smac and preventing it from antagonizing the XIAP-mediated inhibition of caspases, rather than by directly inhibiting caspases Citation[13].
Members of the IAP family contain one or more BIR domains, each of them having different functions. XIAP, NAIP, c-IAP1 and c-IAP2 each contain three BIR domains. In XIAP, the third BIR domain (BIR3) inhibits the activity of caspase 9, whereas the region between BIR1 and BIR2 specifically targets caspase 3 and caspase 7 Citation[14]. Many IAPs also have another protein motif, the RING domain, which can recruit E2 ubiquitin-conjugating enzymes (UBCs) and catalyze the transfer of ubiquitin onto target proteins, leading to proteasomal degradation. Through their ubiquitin E3 ligase activities, IAPs appear to regulate NF-κB family transcriptional activators, which have also been associated with malignancy. c-IAP1 and c-IAP2 promote the degradation of NF-κB-inducing kinase, the central Ser/Thr kinase in the non-canonical NF-κB pathway Citation[15]. cIAP1 and c-IAP2 contain a caspase recruitment domain (CARD) with an unknown role ().
Figure 2. Proteins of the inhibitor of apoptosis family (IAP) include c-IAP1, c-IAP2, XIAP (X-linked IAP), ML-IAP (melanoma IAP/livin), NAIP (neuronal apoptosis inhibitory protein), and survivin. Some of the IAPs contain a protein motif, the RING domain, which can recruit E2 ubiquitin-conjugating enzymes. cIAP1 and c-IAP2 contain a caspase recruitment domain (CARD) with an unknown role.
The activity of IAP family members can be modulated by other endogenous cell factors released by mitochondria. For example, the inhibitory effects of XIAP can be overcome by the antagonizing functions of the mitochondrial proteins, Smac/DIABLO and Omi/HtrA2.
The ubiquitous c-FLIP polypeptide contains a prodomain similar to that of procaspase-8, but lack a caspase active site and may inhibits procaspases 8 and 10.
Extrinsic pathway or death receptors pathway
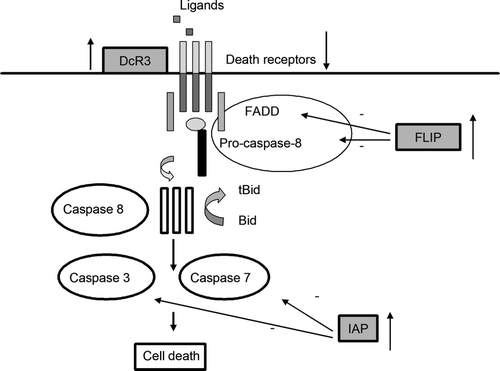
The extrinsic pathway is initiated by stimulation of the transmembrane death receptors by specific ligands released by other cells. Death receptors may belong to the tumor necrosis factor family (TNF), which is composed of many members. Members of the TNF receptor family have a cysteine-rich extracellular subdomain that allows them to specifically recognize their ligands and a cytoplasmic domain of about 80 amino acids called the “death domain” (DD), which plays a critical role in transmitting the death signal from the cell surface to the intercellular pathways.
Currently, six death receptors are known, including TNF receptor 1 (TNF-R1, also called DR1, p55, p60, or CD120a), Fas (also called DR2, CD95, or APO-1), DR3 (also known as Apo-3, LARD, TRAMP or WSL1), TNF-related apoptosis-inducing ligand receptor 1 (TRAIL-R1, also known as DR4 or Apo-2), TRAIL-R2 (DR5, KILLER, or TRICK2), and DR6 Citation[16]. The extrinsic pathway is initiated by binding of the transmembrane death receptors with their specific ligands (FasR/FasL, TNFR1/TNFα, DR-4/TRAIL, DR-5/TRAIL, DR3/Apo-3L/TWEAK). Once activated, the intracellular domains of these receptors (DD) bind to the adaptor protein Fas-associated death domain (FADD) or TRADD (TNFR1-associated death domain protein) to form the death inducing complex (DISC) with recruitment of pro-caspase 8. Next, procaspase 8 is proteolytically activated and serves as the ‘initiator’ caspase, further activating downstream effectors proteins such as caspase 3 and 7 to initiate cell degradation, causing inevitable apoptosis.
Two types of intracellular signaling have been defined for the extrinsic apoptotic pathway that are characterized by the generation of high (type I) or low (type II) levels of DISC and caspase 8 activation upon receptor stimulation. In type I, caspase 8 stimulation is sufficient to activate effector caspases and to induce apoptotic death. In contrast, further amplification is needed in the type II cells, where active caspase 8 cleaves BID protein to tBID, which can bind to pro-apoptotic BAX and BAK, resulting in mitochondrial membrane permeabilization and the release of mitochondrial proteins cytochrome C and DIABLO Citation[17].
Tumor necrosis factor-related apoptosis inducing ligand (TRAIL) can initiate apoptosis by direct caspase activation, as described above, or indirectly via the release of apoptogens from mitochondria. Recent studies suggest that TRAIL induced mitochondrial apoptotic pathway is potentiated by phospholipids scramblase-3 Citation[18]. Phospholipids scramblase (PLS) are enzymes that play an important role in bidirectional movement of membrane lipids, which are critical for mediating apoptosis in many cell types. PLS3 is a phospholipid scramblase from mitochondria that facilitates changes in mitochondrial membrane lipids that promote the release of apoptogenic factors with activation of caspase 9 and effector caspase 3.
The death receptor pathway is regulated at different levels. First, expression of death receptors varies among different cell types at different stage of development. For example, DR4 and DR5 appear to be expressed in the normal adult thymus and in a variety of tumor cells, but not in other normal tissues. The DR3 and DR6 signaling pathways are less characterized. DR3 can co-stimulate T cells, but its role as an apoptosis inducing receptor is less clear Citation[19]. Second, some cells express so-called “decoy receptors”, cell surface or secreted proteins that bind death ligands with high affinity but are unable to transduce signals to cytoplasmic adaptor molecules Citation[20]. Although several decoy receptors of the TNF receptor family have been identified, the most important one involved in many diseases is DcR3. Also known as TR6 or M68, DcR3 is a member of the TNF receptor family and can bind to the Fas ligand to inhibit its ability to induce apoptosis. A third mechanism for inhibiting death receptor signaling involves the down-regulation of procaspase-8. For example, the ubiquitous c-FLIP polypeptide contains a prodomain similar to that of procaspase-8, but lack a caspase active site ().
Figure 3. The extrinsic pathway of apoptosis and its regulation. Death receptor signaling can be inhibited at a different levels, including: overexpression of decoy receptors, diminished expression of death receptors, overexpression of IAP molecules that can inhibit caspase 3 and 7 activity, or overexpression of cellular or viral FLIP molecules that can bind to the death effectors domain of FADD and prevent procaspase 8 recruitment.
In addition, the binding of TRAILR with its ligands can result in the activation of other signaling pathways, including the phosphoinositide 3-kinase (PI3K)–Akt, nuclear factor κB (NFκB) and mitogen-activated protein kinase (MAPK, including extracellular signal-regulated kinase (ERK), Jun N-terminal kinase (JNKs) and p38) pathways. Activation of these additional pathways has a proliferative effect that regulates different physiological processes, such as hematopoiesis and T cell activation and survival, with TRAIL being an important molecule involved in the surveillance and elimination of developing tumors Citation[21].
Intrinsic pathway or the mitochondrial pathway
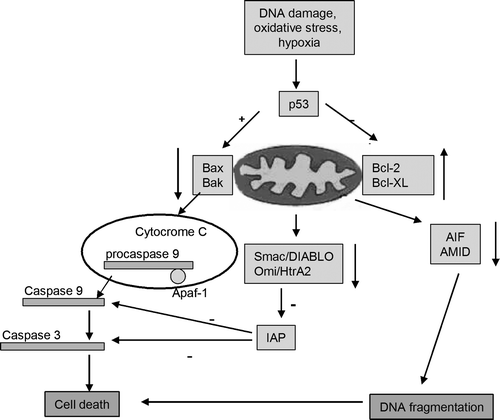
The intrinsic pathway, involving the mitochondria, is usually activated by the loss of growth factor signals or in response to lethal stimuli from inside the cell, such as DNA damage, oxidative stress, hypoxia, or chemotherapeutic drugs.
Mitochondria, very specialized organelles, have an outer membrane (OM) separated from an inner membrane (IM) by an intermembrane space (IMS). The IMS contains many proteins involved in cell death induction, such as cytochrome c (cyt C), apoptosis-inducing factors (AIF), Omi/HtrA2, EndoG and Smac/DIABLO. All of the stimuli that cause changes to the inner mitochondrial membrane produce the opening of the mitochondrial permeability transition pore (MPT), loss of the mitochondrial transmembrane potential and release of sequestered pro-apoptotic proteins from the intermembrane space into the cytosol. After release into the cytoplasm, cytochrome C stimulates apoptosome formation (a complex including apoptotic protease-activating factor [Apaf-1], dATP, cytochrome c and caspase 9) followed by activation of caspase 9. The ‘initiator’ caspase 9 causes the activation of the ‘executioner’ caspases (3, 6, 7), which cleave vital substrates, resulting in cellular death. The catalytic activity of cytochrome C is modulated by members of the inhibitor of apoptosis protein family (IAP), which are in turn controlled by two other mitochondrial proteins, Smac/DIABLO and OMI/HtrA2 Citation[22].
Apoptosis inducing factor (AIF) is a mitochondrial oxidoreductase associated to the inner mitochondrial membrane. During apoptosis AIF can be released by proteolysis and translocate to the nucleus where it participates in chromatin condensation and DNA fragmentation. A newly discovered member of the AIF family is AMID (apoptosis-inducing factor-like mitochondrion-associated inducer of death). Despite its name, its precise cellular localization and its role during apoptosis are unclear. Recent studies in human leukemia Jurkat T-cells suggest increased expression and plasma membrane association of AMID after apoptosis induction Citation[23].
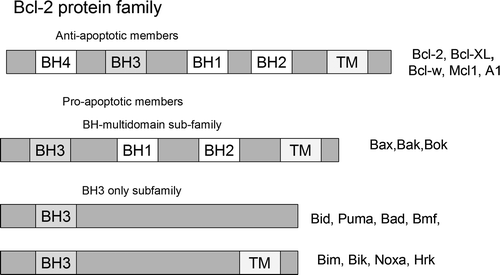
The intrinsic pathway is controlled by interactions between proapoptotic and antiapoptotic members of the Bcl-2 protein family. There are at least 20 proteins in the Bcl-2 family, which are divided into 3 groups. Group I members are anti-apoptotic, whereas groups II and III are pro-apoptotic. The members of the Bcl-2 family can be defined by the presence of conserved sequence motifs known as Bcl-2 homology domains (BH1 to BH4). Each of these BH domains has a different function, and family members contain one or more of them. Whereas most antiapoptotic proteins (including Bcl-2, Mcl-1, Bcl-w, Bcl-xL, and A1) contain all four Bcl-2 homology domains and protect cells exposed to diverse cytotoxic conditions, proapoptotic proteins may lack one or several of the BH domains and can be divided into two subgroups. Members of the first group, BH3-only domain proteins (including Bid, Bim, Bik, Bad, Bmf, Noxa, Puma, and Hrk), act as damage sensors and direct antagonists of Bcl-2 and the other pro-survival proteins. The other proapoptotic group contains the BH 1-3 domains (Bax, Bak, Bok and Bcl-xs) and directly activates other proapoptotic family members. Antiapoptotic Bcl-2 proteins exert their activity by binding the proapoptotic members Bax and Bak, preventing mitochondrial damage Citation[24]. Recently, a novel Bcl-2 protein, named Bcl-xAK, has been identified that contains BH4 and BH2 domains but lacks BH3 and BH1 Citation[25]. The overexpression of Bcl-xAK triggers apoptosis in human melanoma cells. This is the first Bcl-2 protein that induces apoptosis without a functional BH3 domain ().
Figure 4. The extended Bcl-2 family. This family comprises pro-survival proteins, which share the Bcl-2 homology (BH) domains, and pro-apoptotic proteins that contain BH1-3 domains or only the BH3 domain.
The p53 gene is a well-known tumor suppressor gene that encodes a nuclear protein with a critical role in the regulation of cell death. In response to different cellular stresses, p53 stops cell-cycle progression through expression of its target gene, p21. When the cell cannot repair the cellular damage, p53 promotes apoptosis. It seems that p53 uses multiple pathways to induce cell death Citation[26]. It is well established that p53 transactivates a variety of apoptotic factors, such as Bax, Puma, Noxa, and p53-regulated-inducing protein 1 (p53AIP1) and also can translocate to the mitochondria where it binds to the antiapoptotic proteins Bcl-2 and Bcl-xL. In addition, p53 can stimulate the production of reactive oxygen species and Fas/CD95 to redistribute to the cell surface. Therefore, when p53 is inhibited, cell proliferation is accelerated. Indeed, p53 function is compromised in the majority of human cancers.
In normal cells, MDM2 is an essential regulator of p53. MDM2 and p53 regulate each other through an autoregulatory feedback loop. Upon activation, p53 transcribes the MDM2 gene and, in turn, the MDM2 protein inhibits p53 activity. Many models that explain p53 inhibition by MDM2 have been observed. First, MDM2 binds directly to the N-terminal p53 transactivation domain and inhibits its transcriptional activity Citation[27]. Second, MDM2 functions as an E3 ubiquitin ligase, which leads to both the export of p53 to the cytoplasm and its proteasomal degradation. Overexpression of MDM2 due to the amplification of the MDM2 gene has been observed in several human cancers. Targeting MDM2 with small molecules to reactivate p53 has emerged as a promising new cancer therapeutic strategy Citation[26] ().
Figure 5. The intrinsic pathway and its regulation. The mitochondrial pathway can be inhibited at different stages, including: elevated expression of anti-apoptotic Bcl-2 family members, diminished expression of proapoptotic Bcl-2 family members, overexpression of IAP family members that can inhibit caspase 3, caspase 9 and caspase 7 activities, diminished expression of AIF family members or certain molecules expressed in the mitochondria (Smac/Diablo, Omi/HtrA2) that regulate the expression of IAP family members.
Cancer: Mechanisms of apoptosis resistance
Cancer is a genetic disease in which a succession of genetic mutations is observed. It is now believed that some cancers are caused by the lack of cell death, rather than an increased rate of proliferation.
Transformed cells are recognized by the immune system based on the expression of abnormal molecules on the cell surface and the abnormal behavior of preneoplastic cells that respond to oncogenic stress. The immune system distinguishes between normal programmed cell death (PCD), which appears in development and homeostasis, and pathogen-induced PCD. This is possible using specific receptors expressed on the surface or within the cytoplasm of innate immune effectors that recognize so-called pathogen-associated molecular patterns (PAMPs). In addition to PAMPs, the so-called “danger-associated molecular patterns” can trigger the immune response. Efficient recognition of transformed cells and their elimination is called “immunosurveillance” Citation[28]. Transformed cells often escape from immunosurveillance due to their capacity to hide the abnormal molecules present on their surface by targeting specific pro-survival strategies or by inhibiting apoptosis pathways Citation[29]. In tumorigenesis, disturbance of both extrinsic and intrinsic pathways of apoptosis is involved.
The death receptor pathway is regulated, as specified above, at three different levels. All of the levels can be disturbed in cancer cells. Concerning the death receptors, decreased expression of the Fas receptor has been observed in hepatomas as compared to normal hepatocytes, and its downregulation might contribute to evasion of the immune system during liver carcinogenesis Citation[30]. In addition, DR4 and DR5 appear to be expressed in a variety of tumor cells, but not in normal tissues. Second, several decoy receptors of the TNF receptor family have been identified, the most important one being DcR3. DcR3 is over expressed in a variety of human cancers, such as colon, lung, and stomach cancer. Some studies suggest that DcR3 can be used to predict lymph node invasion in patients with gastric cancer Citation[31]. A third mechanism for inhibiting death receptor signaling involves the downregulation of procaspase 8 by specific molecules such as c-FLIP. Overexpression of c-FLIP is observed in many carcinomas, with interruption of the death signal by its binding to FADD and competitive inhibition of the recruitment of procaspases 8 and 10 Citation[32].
The intrinsic pathway is controlled by interactions between proapoptotic and antiapoptotic members of the Bcl-2 protein family. Alteration in the expression of either type occurs frequently in many human cancers. Bcl-2 is overexpressed in a variety of cancers and was the first apoptosis-related gene recognized to play a role in tumorigenesis. Elevations in Bcl-2 protein levels are commonly found in many hematopoietic malignancies including multiple myeloma (MM), acute lymphocytic leukemia (ALL), and chronic lymphocytic leukemia (CLL) Citation[33]. Furthermore, elevations in Mcl-1 have been reported in acute leukemia after relapse from chemotherapy Citation[34]. Mutated or downregulated Bax and Bak are also observed in certain cancers Citation[35].
Inhibitors of apoptosis proteins (IAPs) are overexpressed in many human cancers, and their level of expression has been associated with treatment resistance. For example, the overexpression of c-IAP1, c-IAP2, NAIP, XIAP and survivin has been detected in breast cancer Citation[36]. In addition c-IAP2 is overexpressed in low and high grade pancreatic intraepithelial neoplastic lesions and pancreatic ductal adenocarcinomas. Thus, it is considered an early event in the progression of pancreatic cancer Citation[37]. XIAP is overexpressed in esophageal cancer tissues compared with normal tissues and might confer resistance to apoptosis induction by caspase 3 activation and promote tumorigenesis Citation[38].
Half of all human cancers have mutations in the tumor-suppressor gene p53, considered to be the “guardian of the genome”. In human cancer, the function of p53 is inhibited by its primary cellular inhibitor, MDM2. Overexpression of MDM2 due to amplification of the MDM2 gene has been observed in many human cancers. Since p53 plays a central role in apoptosis induction, alteration of the p53 pathway influences the sensitivity to apoptosis and modifies the body homeostasis.
The nuclear factor-kappa B (NF-κB) pathway is an important signaling pathway in tumorigenesis which regulates tumor cell proliferation, controls apoptosis, promotes angiogenesis, and stimulates invasion and metastasis. Due to its multiple roles in tumorigenesis, inhibition of (NF-κB) alone or in combination with chemotherapy may lead to growth inhibition or tumor cell death Citation[39].
Cancer: Target therapy involving the apoptotic pathway
The greatest problem limiting effective cancer treatment may be drug resistance which can involve multiple mechanisms including influx or efflux of drugs, resistance to death stimuli, or activation of proliferation pathways. Strategies to overcome tumor resistance to either extrinsic or intrinsic apoptotic pathways are under investigation. These include activation of the extrinsic pathway through proapoptotic receptors, restoration of p53 activity, inhibition of the Bcl-2 family of proteins, BH3-only mimic proteins, caspase modulation, IAP inhibition, and proteasome inhibition.
Targeting pro-apoptotic receptors in cancer
The potential targeting the extrinsic apoptotic signaling pathway as a therapeutic strategy was revealed more than 15 years ago when Trauth and colleagues demonstrated that a single intravenous injection of an APO-1 antibody induced regression of a xenotransplanted human B-cell tumor in nude mice within a few days Citation[40]. The TRAIL pathway is an attractive therapeutic target for cancer treatment because tumor cells are more sensitive to the TRAIL-dependent apoptosis pathway than are normal cells. While the pro-apoptotic ligand FasL has cytotoxic activity against many tumor cells, hepatic toxicity limits the use of Fas-targeted therapy Citation[41]. Two classes of pro-apoptotic receptors antagonists (PARAs) that target DR4 or DR5 have been developed. Recombinant human (rh) Apo2L/TRAIL activates both receptors while monoclonal antibodies have been developed that act as agonists of either DR4 (mapatumumab) or DR5 (lexatumumab, Apomab, AMG655, Cs-1008, LBY-135) Citation42–46. Stimulation of the proapoptotic receptors DR4 and DR5 shows promising safety and efficacy in several diverse preclinical cancer models. These agents have shown minimal cytotoxicity of normal cells and have been demonstrated to kill cells that are resistant to standard chemotherapy or restore the sensitivity of tumor cells to chemotherapy when combined with standard treatment. The most common adverse events that have been reported include fever, fatigue, thrombocytopenia, and manageable digestive toxicities. Moreover, several of these agents have successfully met the safety criteria of phase I clinical trials. Phase II trials are being initiated to assess the safety and efficacy of specific PARAs both in combination with standard cytotoxic chemotherapy or with targeted agents in a variety of tumors types. Because cancer cells are not universally sensitive to treatment, there has been extensive research to identify and characterize potential biomarkers for sensitivity to Apo2/TRAIL. Recent studies have identified the myc oncogene and O-glycosylation enzymes as potential markers for Apo2/TRAIL sensitivity in cancer cells Citation[47], Citation[48].
Targeting intracellular caspase inhibitors
Members of the IAP family have been investigated as therapeutic targets for the treatment of multiple diseases including cancer due to the overexpression of its members in many tumors. There are currently several preclinical studies involving the administration of small molecule drugs that mimic the activity of Smac by binding to IAPs and preventing their interaction with caspases or use antisense-based inhibitors of IAPs Citation[49], Citation[50]. Another possibility to deplete IAP expression is a molecular method named RNA interference (RNAi), which consists of synthesizing and transferring small interfering RNA (siRNA) into cancer cells to block the overexpression of IAPs or other molecules. Treatment with XIAP siRNA in combination with chemotherapy can efficiently decrease XIAP expression and induce cellular apoptosis Citation[38]. On the other hand, normal cells are less dependent on IAPs, thus providing an important advantage for these novel agents. FLIP [FLICE (FADD-like ICE)-inhibitory protein] is an endogenous inhibitor of extrinsic apoptosis signaling. Recently, two compounds that reduce the expression of c-FLIP and sensitize cells to Apo2L/TRAIL-induced apoptosis have been described Citation[51].
Targeting the p53-MDM2 interaction
Small-molecule inhibitors of the MDM2-p53 interaction have been developed. The first ones, called the nutlins, were reported in 2004, but in the last four years several new classes of small molecule MDM2 inhibitors have been discovered, including Nutlin-3, MI-219, and MI-63 Citation[52], Citation[53]. The nutlins have been shown to inhibit tumor growth by inducing cell cycle arrest and cell death. In several xenograft models of human cancer nutlin-3 and MI-219 have shown strong antitumor activity in the presence of wild-type p53, but lack activity against tumors deficient in wild-type p53 Citation[53]. The wild-type status of p53 appears to be the major determinant of the antitumor activity of MDM2 inhibitors. In addition, it has recently been shown that nutlin 3 can downregulate TNFα-induced activation of the NF-?B reporter genes in lung cancer cells Citation[54]. Importantly, during treatment with these small molecules, no visible signs of toxicity in the animals were observed as assessed by necropsy studies and antibody weight. MDM2 inhibitors are promising therapeutic compounds with several small molecules now in preclinical development. Because the activity of MDM-2 is p53 dependent and 50% of tumors contain p53 mutations, it is reasonable to identify a biomarker of p53 activity. A member of the transforming growth factor β superfamily called macrophage inhibitory cytokine-1 has been proposed as a biomarker for p53 activity Citation[55].
Targeting Bcl-2 family proteins
Bcl-2 is overexpressed in a variety of cancers and was the first apoptosis-related gene recognized to play a role in tumorigenesis. Treatment of tumors with Bcl-2 anti-sense oligonucleotides can reduce tumor growth and clinical trials have shown promising preliminary results including improvements in the response rate and progression free survival in patients with chronic lymphocytic leukemia and advanced, relapsed melanoma Citation[56], Citation[57]. RNAi may also be a useful method by which to downregulate the expression of anti-apoptotic genes; several small molecule antagonists of Bcl-2 or Bcl-XL are in early phases of development Citation[58]. Promising results have been reported in lung cancer, where in combination with chemotherapy, it was possible to sensitize resistant cell lines and to induce apoptosis Citation[59]. Another strategy to enhance the response of apoptotic stimuli would be to stimulate the expression of pro-apoptotic molecules. This approach is under investigation Citation[60].
There is now great interest in developing drugs that mimic the action of the BH3 domain by binding to one or more of the Bcl-2 like proteins and triggering apoptosis. Selective interactions of the BH3-only proteins with pro-survival molecules are now well documented Citation[61]. Several short peptides that could be used either as tools to understand the roles of the Bcl-2 family of proteins or to develop a new generation of anticancer drugs have been developed. One of these is GX15-070 which binds to Bcl-2, Bcl-XL, Bcl-w and Mcl1 and has activity in multiple cancer cell lines tested Citation[62]. AT-101 has cytotoxic potency similar to that of GX15-070, and was effective in a phase I open-label trial in patients with previously untreated chronic lymphocytic leukemia. This compound had manageable gastrointestinal toxicities and has been tested in phase II studies in combination with rituximab with promising results Citation[63]. The other two drugs: ABT-737 and ABT-263 bind to Bcl-2, Bcl-xL and Bcl-w, but not with Mcl-1 or A1, and induced stable regression of both human lung cancer and hematological disease in mouse xenograft models with minimal effects on platelet counts Citation[64], Citation[65]. In addition, ABT-737 can enhance TRAIL-mediated cytotoxicity by unsequestering Bim and Bak in human pancreatic cancer cell lines Citation[66].
Targeting protein degradation: E1 inhibitors
Protein degradation plays a vital role in controlling the activity of key molecules involved in cell cycle progression and apoptosis, including the p53 and NK-κB pathways. Recently, great advances have been made in our understanding of the fundamental importance of the ubiquitin-proteasome pathway in diverse biological processes. Ubiquitin-activating enzymes, also known as E1 enzymes, catalyze the first step in the ubiquitination process that targets a protein for degradation via the proteasome. Alterations in ubiquitination are observed in a wide range of pathological conditions, including cancer. Several classes of proteasome inhibitors that block degradation of ubiquitinylated proteins have been developed Citation[67]. Bortezomid, approved by the FDA for the treatment of relapsed and refractory multiple myeloma, has promising anticancer activity in multiple other tumor types by effecting cellular proliferation, survival, apoptosis, and angiogenesis Citation[68]. Early phase I and II trials have suggested activity for bortezomid in lung cancer Citation[69].
PYR-41 is another proteasome inhibitor that increases the level and activity of p53, as well as the levels of MDM2 and p21, another p53 target gene. PYR-41 can increase apoptosis in cells with wild-type p53 and works by inhibiting several key steps of the NK-κB pathway. PYR-41 inhibits both proteasome-dependent and proteasome-independent ubiquitinylation, but its molecular mechanism needs to be more fully elucidated Citation[67].
Single agent therapy may be more likely to result in the development of resistant tumors, a finding thought to be less common with the use of combinations of drugs. The ligation of TRAILRs in cells that cannot initiate an apoptotic signal results in activation of the NK-κB and PI3K-Akt pathways with stimulation of cell proliferation, survival, and cell migration. This provides a scientific rationale to combine agents that target the TRAIL pathway with additive effects on apoptosis ()Citation70–77.
Table 1. Effect of various agents on Apo2/TRAIL in preclinical studies.
The examples mentioned represent some of the current research in the apoptosis field and provide examples of the use of specific target therapy in improving the outcome of anti-cancer therapy.
Conclusion
Identifying the key proteins involved in apoptosis represents an attractive way to prevent the development of many diseases including cancer. Understanding how these proteins affect the apoptotic pathways may lead to more effective cancer treatments. Research efforts over the past decade have contributed to our knowledge regarding the characterization of the molecules involved in apoptosis, the relationships between these molecules, and the key molecules that can be used in targeted therapy.
The discovery of apoptosis pathways and the development of specific molecules that induce apoptosis of tumor cells suggest that cell death can be targeted therapeutically. However, the success of pro-apoptotic therapies has been limited, perhaps because of our lack of understanding the complexity of cell death regulation.
Acknowledgements
We gratefully acknowledge members of Functional Genomics and Experimental Pathology from Cancer Institute “I Chiricuta” Cluj-Napoca for helpful comments and suggestions.
References
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 2009; 16: 3–11
- Vanlangenakker N, Berghe TV, Krysko DV, Festjens N, Vandenabeele P. Molecular mechanisms and pathophysiology of necrotic cell death. Curr Mol Med 2008; 8: 207–20
- Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature 2000; 407: 784–8
- Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA. Induction of immunological tolerance by apoptotic cell requires caspase-dependent oxidation of high-mobility group Box-1 protein. Immunity 2008; 29: 1–2
- Birge RB, Ucker DS. Innate apoptotic immunity: The calming touch of death. Cell Death Differ 2008; 15: 1096–102
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nature Rev Mol Cell Biol 2007; 8: 741–52
- Ullman E, Fan Y, Stawowczyk M, Chen H-M, Yue Z, Zong W-X. Autophagy promotes necrosis in apoptosis-deficient cells in response to ER stress. Cell Death Differ 2008; 15: 422–5
- Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117: 326–36
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10: 51–64
- Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, Vandenabeele P. Caspases in cell survival, proliferation and differentiation. Cell Death Differ 2007; 14: 44–55
- Kang SJ, Wang S, Kuida K, Yuan J. Distinct downstream pathways of caspase-11 in regulating apoptosis and cytokine maturation during septic shock response. Cell Death Differ 2002; 9: 1115–25
- Deveraux QL, Reed JC. IAP family proteins-suppressors of apoptosis. Genes Dev 1999; 13: 239–52
- Vucic D, Franklin MC, Wallweber HJ, Das K, Eckelman BP, Shin H, et al. Engineering MLIAP to produce an extraordinarily potent caspase 9 inhibitor: Implications for Smac-dependent anti-apoptotic activity of ML-IAP. Biochem J 2005; 385: 11–20
- Vaux DL, Silke J. IAPs, RINGs and ubiquitylation. Mol Cell Biol 2005; 6: 287–97
- Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 2007; 131: 669–81
- Suliman A, Lam A, Datta R, Srivastava RK. Intracellular mechanisms of TRAIL: Apoptosis through mitochondrial-dependent and independent pathways. Oncogene 2001; 20: 2122–33
- Ozoren N, El-Deiry WS. Defining characteristics of Types I and II apoptotic cells in response to TRAIL. Neoplasia 2002; 4: 551–7
- Ndebele, K, Gona, P, Jin, TG, Benhaga, N, Chalah, A, Degli-Esposti, M, , et al. Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) induced mitochondrial pathway to apoptosis and caspase activation is potentiated by phospholipids scramblase-3. Apoptosis 2008;13.
- Meylan T, Davidson TS, Kahle E, Kinder M, Acharya K, Jankovic D, Bundoc V, et al. The TNF-family receptor DR3 is essential for diverse T cell-mediated inflammatory diseases. Immunity 2008; 29: 79–89F
- Ashkenazi A, Dixit VM. Apoptosis control by death and decoy receptors. Curr Opin Cell Biol 1999; 11: 255–60
- Falschlehner C, Emmerich CH, Gerlach B, Walczak H. TRAIL signalling: Decisions between life and death. Int J Biochem Cell Biol 2007; 39: 1462–75
- Cory S, Adam JM. The Bcl-2 family: Regulators of the cellular life-or-death switch. Nat Rev Cancer 2002; 2: 647–56
- Bilyy, R, Kitl, Y, Hellman, U, Stoika, R. AMID: New insights on its intracellular localization and expression at apoptosis. Apoptosis 2008;13.
- Scorrano L, Korsmeyer SJ. Mechanisms of cytochrome c release by proapoptotic BCL-2 family members. Biochem Biophys Res Commun 2003; 304: 437–44
- Hossini M, Geilen CC, Fecker LF, Daniel PT, Eberle J. A novel Bcl-x splice product, Bcl-xAK triggers apoptosis in human melanoma cells without BH3 domain. Oncogene 2006; 25: 2160–9
- Dey A, Verma CS, Lane DP. Updates on p53: Modulation of p53 degradation as a therapeutic approach. Br J Cancer 2008; 98: 4–8
- Ringshausen I, O'Shea CC, Finch AJ, Swigart LB, Evan GI. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell 2006; 10: 501–14
- Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L, et al. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ 2008; 15: 3–12
- Ullrich E, Bonmort M, Mignot G, Kroemer G, Zitvogel L. Tumor stress, cell death and the ensuing immune response. Cell Death Differ 2008; 15: 21–8
- Hiroshige N, Mitsuo N. The alteration of Fas receptor and ligand system in hepatocellular carcinoma – How do hepatoma cells escape from the host immune surveillance in vivo?. AINO J 2004; 3: 61–6
- Wu Y, Guo E, Yu J, Xie Q. High DcR3 expression predicts stage pN2-3 in gastric cancer. Am J Clin Oncol 2008; 31: 79–83
- Li X, Pan X, Zhang H, Lei D, Liu D, Xu F, et al. Overexpression of cFLIP in head and neck squamous cell carcinoma and its clinicopathologic correlations. J Cancer Res Clin Oncol 2008; 134: 609–15
- Pepper C, Bentley P, Hoy T. Regulation of clinical chemoresistance by Bcl-2 and Bax oncoproteins in B-cell chronic lymphocytic leukaemia. Br J Haematol 1996; 95: 513–7
- Inoue S, Walewska R, Dyer MJS, Cohen GM. Downregulation of Mcl-1 potentiates HDACi-mediated apoptosis in leukemic cells. Leukemia 2008; 22: 819–25
- Kondo S, Shinomura Y, Miyazaki Y, Kiyohara T, Tsutsui S, Kitamura S, et al. Mutation of the bax gene in human gastric and colorectal cancers. Cancer Res 2000; 60: 4328–30
- Peng XH, Karna P, O'Regan RM, Liu X, Naithani R, Moriaty R, et al. Down-regulation of inhibitor of apoptosis proteins by deguelin selectively induces apoptosis in breast cancer cells. Mol Pharmacol 2007; 71: 101–11
- Esposito I, Kleeff J, Abiatari I, Shi X, Giese N, Bergmann F, et al. Overexpression of cellular inhibitor of apoptosis protein 2 is an early event in the progression of pancreatic cancer. J Clin Pathol 2007; 60: 885–95
- Zhang S, Ding F, Luo A, Chen A, Yu Z, Ren S, et al. XIAP is highly expressed in esophagean cancer and its downregulation by RNAi sensitizes esophageal carcinoma cell lines to chemotherapeutics. Cancer Biol Ther 2007; 6: 973–80
- Richmond A. Nf-Kappa B, chemokine gene transcription and tumor growth. Nat Rev Immunol 2002; 2: 664–74
- Trauth BC, Klas C, Peters AM, Matzku S, Moller P, Falk W, et al. Monoclonal antibody mediated tumor regression by induction of apoptosis. Science 1989; 245: 301–5
- Lawrence D, Shahrokh Z, Marsters S, Achilles K, Shih D, Mounho B, et al. Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions. Nat Med 2001; 7: 383–5
- Herbst RS, Mendolson DS, Ebbinghaus S, Gordon MS, O'Dwyer P, Lieberman G, et al. A phase I safety and pharmacokinetics study of recombinant Apo2L/TRAIL an apoptosis inducing protein in patients with advanced cancer. Proc Am Soc Clin Oncol 2006; 24, Abstract 301
- Tolcher AW, Mita M, Meropol NJ, Mehren M, Patnaik A, Padavic K, et al. Phase I pharmacokinetic and biologic correlative study of mapatumumab, a fully human monoclonal antibody with agonist activity to tumor necrosis factor-related apoptosis-inducing ligand receptor-1. J Clin Oncol 2007; 25: 1390–5
- Camidge DR, Herbst RS, Gordon M, Eckhardt S, Kurzroc R, Durbin B, et al. A phase I safety and pharmacokinetic study of apomab, a human DR5 agonist antibody, in patients with advanced cancer. J Clin Oncol 2008, Suppl 18: 3582, Abstract 25
- Sharma S, de Vries EG, Infante JR, Oldenhuis C, Chiang L, Bilic S, et al. Phase I trial of LBY135, a monoclonal antibody agonist to DR5, alone and in combination with capecitabine in advanced solid tumors. J Clin Oncol 2008, Suppl: 3538, Abstract 26
- Saleh MN, Percent I, Wood TE, Posey J, Shah J, Carlisle R, et al. A phase I study of CS-1008 (humanized monoclonal antibody targeting death receptor 5 or DR5) administered weekly to patients with advanced slid tumors or lymphomas. J Clin Oncol 2008, Suppl: 3537, Abstract 26
- Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med 2007; 13: 1070–7
- Ricci MS, Jin Z, Dews M, Yu D, Thomas-Tikhonenko A, Dicker DT, et al. Direct repression of FLIP expression by myc is a major determinant of TRAIL sensibility. Mol Cell Biol 2004; 24: 8541–55
- Schimmer AD, Welsh K, Pinilla C, Wang Z, Krajewska M, Bonneau MJ, et al. Small-molecules antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer Cell 2004; 5: 25–35
- LaCasse EC, Cherton-Horvat GG, Hewitt KE, Jerome LJ, Morris SJ, Kandimalla ER, et al. Preclinical characterization of AEG35156/GEM 640 a second-generation oligonucleotide targeting X-linked inhibitors of apoptosis. Clin Cancer Res 2006; 12: 5231–41
- Schimmer AD, Thomas MP, Hurren R, Gronda M, Pellecchia M, Pond GR, et al. Identification of small molecules that sensitize resistant tumor cells to tumor necrosis factor family death receptors. Cancer Res 2006; 66: 2367–75
- Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med 2007; 13: 23–31
- Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA 2008; 105: 3933–8
- Dey A, Wong ET, Bist P, Tergaonkan V, Lane David P, et al. Nutlin-3 inhibits the NF-κB pathway in a p53-dependent manner: Implication in lung cancer. Ther Cell Cycle 2007; 6: 2178–85
- Yang H, Filipovic Z, Brown D, Breit SN, Vassilev LT. Macrophage inhibitory cytokine-1: A novel biomarker for p53 pathway activation. Mol Cancer Ther 2003; 2: 1023–9
- O'Brien S, Moore JO, Boyd TE. Randomized phase II trial of fludarabine plus cyclophosphamide with or without oblimersen sodium (Bcl-2 antisense) in patients with relapsed or refractory chronic lymphocytic leukaemia. J Clin Oncol 2007; 25: 1114–20
- Bedikian AY, Millward M, Pehamberger H, Conry R, Gore M, Trefzer U, et al. Bcl-2 antisense (olimersen sodium) puls dacarbazine in patients with advanced melanoma: The Oblimersen Melanoma Study. J Clin Oncol 2006; 24: 4738–45
- Trudel S, Li ZH, Rauw J, Tiedemann RE, Wen XY, Stewart AK, et al. Preclinical studies of the pan-Bcl inhibitor obatolax (GX015-070) in multiple myeloma. Blood 2007; 109: 5430–8
- Huang Z, Lei X, Zhong M, Zhu B, Tang S, Liao D. Bcl-2 small interfering RNA sensitizes cisplatin-resistant human lung adenocarcinoma A549/DDp cell to cisplatin and diallyl disulfide. Acta Biochim Biophys Sin 2007; 39: 835–43
- Hioki M, Kagawa S, Fujiwara T, Sakai R, Kojima K, Watanabe Y, et al. Combination of oncolytic adenovirotherapy and Bax gene therapy in human cancer xerografted models. Potential merits and hurdles for combination therapy. Int J Cancer 2008; 122: 2628–33
- Chen L, Willis S, Wei A, Smith B, Fletcher J, Hinds M, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005; 17: 393–403
- Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Estrov Y, et al. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimic GX15-070 (obatoclax). Cancer Res 2008; 68: 3413–20
- Castro JE, Loria OJ, Aguillon RA, et al. A phase II, open label study of AT101 in combination with rituximab in patients with relapsed or refractory chronic lymphocytic leukemia. Evaluation of two dose regimens. Blood 2007; 110: 917A–918A
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 2008; 68: 3421–8
- Vogler M, Dinsdale D, Sun XM, Young KW, Butterworth M, Nicotera P, et al. A novel paradigm for rapid ABT-737-induced apoptosis involving outer mitochondrial membrane rupture in primary leukaemia and lymphoma cells. Cell Death Differ 2008; 15: 820–30
- Huang S, Sinicrope FA. BH3 mimetic ABT-737 potentiates TRAIL-mediated apoptotic signaling by unsequestering Bim and Bak in human pancreatic cancer cells. Cancer Res 2008; 68: 2944–51
- Voorhees PM, Dees EC, O'Neil B, Orlowski RZ. The proteasome as a target for cancer therapy. Clin Cancer Res 2003; 9: 6316–25
- Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005; 352: 2487–98
- Davies, AM, Lara, PN, Jr, Mack, PC, Gandara, DR. Incorporating bortezomib into the treatment of lung cancer. Clin Cancer Res 2007;13(15 Pt 2):s4647–s4651.
- Ashkenazi A, Pai PC, Fong S, Leung S, Lawrence DA, Marsters SA, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest 1999; 104: 155–162
- El-Zawahry A., McKillop J, Voelkel-Johnson C. Doxorubicin increases the effectiveness of Apo2L/TRAIL for tumor growth inhibition of prostate cancer xenografts. BMC 2005; 5: 2
- Shankar S, Singh TR, Chen X, Thakkar H, Firnin J, Srivastava RK, et al. The sequential treatment with ionizing radiation followed by TRAIL/Apo2L reduces tumor growth and induces apoptosis of breast tumor xenografts in nude mice. Int J Oncol 2004; 24: 1133–40
- Ray S, Bucur O, Almasan A. Sensitization of prostate carcinoma cells to Apo2L/TRAIL by a Bcl-2 family protein inhibitor. Apoptosis 2005; 10: 1411–8
- Brooks AD, Ramirez T, Toh U, Onksen J, Elliott P, Murphy WJ, et al. The proteasome inhibitor bortezomib (Velcade) sensitizes some human tumor cells to Apo2L/TRAIL-mediated apoptosis. Ann NY Acad Sci 2005; 1059: 160–7
- Guo F, Nimmanapalli R, Paranawithana S, Wittman S, Griffin D, Bali P, et al. Ectopic overexpression of second mitochondria-derived activator of caspases (Smac/DIABLO) or cotreatment with N-terminus of Smac/DIABLO peptide potentiates epothilone B derivative-(BMS 247550) and Apo-2L/TRAILinduced apoptosis. Blood 2002; 99: 3419–26
- Fulda S, Wick W, Weller M, Debatin KM. Smac agonists sensitize for Apo2L/TRAIL – or anticancer drug-induced apoptosis and induce regression of malignant glioma in vivo. Nat Med 2002; 8: 808–15
- Secchiero P, Zerbinati C, di Iasio MG, Melloni E, Tiribelli M, Grill V, et al. Synergistic cytotoxic activity of recombinant TRAIL plus the non-genotoxic activator of the p53 pathway nutlin-3 in acute myeloid leukemia cells. Curr Drug Metab 2007; 8: 395–403