
To the Editor,
Lynch syndrome is the most common hereditary colorectal cancer (CRC) syndrome [Citation1]. It is an autosomal dominant condition with mutations in the DNA mismatch repair (MMR) genes (mostly MLH1, MSH2, MSH6 and PMS2) and in the EPCAM gene. Thus repair of DNA mismatch is hindered. High risk of CRC and gynecological malignancies is characteristic of the syndrome. The risk is also higher for gastric, small bowel, biliopancreatic and urinary tract cancers as well as for brain tumors [Citation2].
The offspring of the parents both of whom suffer from Lynch syndrome have a 25% probability of inheriting mutated MMR genes from both parents. The condition is known as constitutional mismatch repair deficiency (CMMRD). It develops in individuals harboring homozygous or biallelic MMR gene mutations. It means that the parents must have mutations in the same MMR gene MLH1, MSH2, MSH6 or PMS2. CMMRD is a rare condition with less than 200 patients reported since the first diagnosis in 1999 [Citation3]. Due to biallelic MMR gene defect, these individuals are prone to develop various malignancies from early age. The spectrum includes different hematological malignancies and central nervous system tumors, as well as Lynch syndrome-associated cancers [Citation4]. Besides, many patients with CMMRD have multiple café au lait spots resembling neurofibromatosis type 1. During their lifetime they usually suffer from more than one malignancy. Hematological malignancies and brain tumors are largely diagnosed in the first decade of life, CRC in the second decade, and other tumors in young adulthood [Citation4].
Case report
In October 2014, a 17-year-old female presented to the surgical oncology outpatient department of Tartu University Hospital, Clinic of Hematology and Oncology. The patient complained of rectal bleeding after defecation and occasionally also without defecation. During recent months she had had an urge to defecate several times a day and the feeling of not being able to completely empty the bowel. She had passed rectoscopic investigation at which a number of polyps had been found in the rectum. The biopsies had revealed adenomas with high-grade dysplasia.
Previously, at the age of 10 in 2008, the patient had been diagnosed and treated for acute T-lymphoblast leukemia. Chemotherapy was administered until November 2010 when the patient was 12. She has been in full remission since then.
Further, neurofibromatosis was suspected considering a number of café au lait spots on the patient's skin as well as recently diagnosed neurofibromatosis in her half-brother.
Proceeding from the complicated anamnesis and clinical findings, the surgical oncologist sent the patient for colonoscopy, upper GI endoscopy and genetic counseling for suspected familial adenomatous polyposis (FAP).
The genetic consultation took place in 2014 where a thorough family history was taken. The patient was the first child born to the non-consanguine parents. The family had no cases of colorectal polyposis in their history. There were some malignancy cases in the proband’s family but the Amsterdam criteria for Lynch syndrome were not fulfilled. In the mother’s family there were two people with malignancies: the grandmother had been diagnosed with leukemia at the age of 75 years and a great-uncle with prostate cancer at the age of 70 years. In the father’s family there were three cancer cases: the grandmother and a great-aunt had had breast cancer at the age of 45–50 years; and the son of the latter had been diagnosed with CRC at the age of 55 years. The proband’s pedigree is shown in .
Figure 1. Proband’s pedigree. BC: breast cancer; CLL: chronic lymphocytic leukemia; CRC: colorectal cancer; PC: prostate cancer. Numbers designate person’s age, age at death and age at cancer diagnosis.
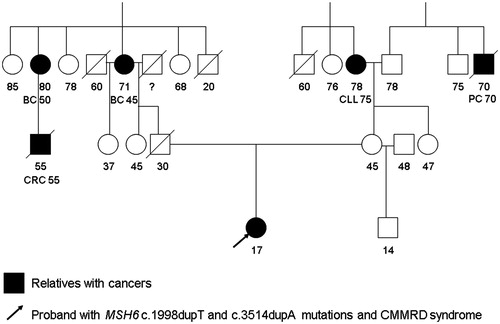
Taking into account the family history, anamnesis and physical examination, the geneticist suspected CMMRD. We investigated DNA (extracted from blood) by new generation sequencing on the Illumina TruSeq Cancer panel using the MiSeq Desktop Sequencer platform. The proband’s DNA analysis revealed two heterozygous mutations in the MSH6 gene: NM_000179.2:c.3514dupA (p.Arg1172Lysfs*5) and NM_000179.2:c.1998dupT (p.Asp667*). The c.3514dupA mutation is described in 14 patients as pathogenic in the InSiGHT database. The c.1998dupT is a new, previously undescribed mutation. According to the Mutation Taster program, this mutation was detected as pathogenic.
The genetic test showed that the patient’s mother and half-brother were carriers of the MSH6 c.1998dupT mutation. Both passed colonoscopy and their colon mucosa was found to be normal.
As the proband’s father had perished in a car accident at the age of 30 years his genetic tests were not available. However, he must have been a carrier of MSH6 c.3514dupA mutation as we found the same mutation in his sister’s DNA. Consequently, the clinical diagnosis of CMMRD of the patient was confirmed by genetic investigation. She underwent colonoscopy. Polyps started right at the dentate line of the anal canal. A number of polyps of different sizes (up to 3 cm in diameter) and shapes were found in the rectum, sigmoid colon and cecum. There was no normal mucosa around the polyps in the above regions. It was all covered with tiny polypoids. Patches of normal looking mucosa were seen in the ascending colon, transverse colon and descending colon, but many of the folds were again covered with tiny polyp-like lesions.
Pathological examination of the biopsies from different polyps and of the surrounding mucosa with tiny polypoids in the rectum revealed tubulovillous adenomas with high-grade dysplasia in all biopsies except for one low-grade dysplasia in one biopsy set.
Immunohistochemical analysis of the patient’s colorectal adenoma tissue as well as normal tissues for MMR protein expression showed no expression of MSH6 and MSH2 proteins; the expressions of MLH1 and PMS2 proteins were positive.
Upper GI endoscopy was performed, together with colonoscopy, under general anesthesia. There were no pathological findings in the esophagus, stomach or duodenal bulb. The papilla of Vater was normal. There was a 5 mm umbilicated flat lesion in the descending duodenum. Pathological examination showed signs of low-grade dysplasia at the border of the biopsies, indicating a possibility of the presence of a small tubular adenoma.
Magnetic resonance imaging (MRI) of the brain did not reveal any changes of tumorous character.
The patient was discussed at a meeting of the oncological multidisciplinary team. It was taken into account that she had CMMRD, with most of the colon and the whole of the rectum being covered with adenomas and an aberrant mucosa. The tubulovillous character of the adenomas of which almost all displayed high-grade dysplasia, was considered. Therefore, total proctocolectomy was proposed.
Two modalities of the operation, total proctocolectomy with end ileostomy, or the same procedure with the ileo-anal anastomosis, were discussed with the patient and her mother. Both of them were consulted by a palliative care specialist and both passed practical training in coping with a stoma and stoma bag. Considering the fact that the adenomas started at the anal canal, but also the uncertain functional results of the ileo-anal anastomosis and the relative ease of using stoma bags, they decided in favor of the operation completed with end ileostomy.
Laparoscopic total proctocolectomy with end ileostomy was performed in July 2015 and it was uneventful. The removed specimen consisted of an ileum segment of 9 cm, the whole colon and rectum together with the anal canal. Macroscopically, multiple polyps started at the dentate line of the anal canal. Throughout the length up to the splenic flexure there were 17 sessile and pedunculated polyps with a diameter of 10–50 mm and 11 polyps with a diameter of 5–9 mm. There were numerous small 2–5 mm sessile polyps in the right side of the colon and a 30 mm sessile polyp in the cecum.
Microscopically, the polyps were mostly tubular adenomas exhibiting high-grade dysplasia. In the biggest polyp (50 mm) from the sigmoid colon, a focus of intraepithelial adenocarcinoma (pTis) was found. Small sessile polyps were found to be early adenomas.
The patient was discharged on the 10th post-operative day. The delay was mainly due to a long-term pain syndrome which was probably associated with emotional lability.
A surveillance program according to a protocol proposed by the European Consortium ‘Care for CMMR-D’ [Citation3] was set up for the patient:
hematological follow-up with a 6-month interval;
brain MRI with a 6–12-month interval;
upper gastrointestinal endoscopy and video capsule endoscopy annually;
gynecological examination, transvaginal ultrasound, endometrial sampling and urine cytology annually.
Discussion
The CMMRD is a disastrous syndrome as most carriers suffer from more than one malignancy since childhood and its prognosis is generally poor. Due to the infrequency of the syndrome, there is a lack of relevant knowledge among the medical community. The uncertain phenotype, various forms of clinical presentation and often a vague hereditary pattern hinders further its recognition by doctors. As a result, the condition is underdiagnosed.
In our case there was no apparent family history raising suspicion of Lynch syndrome. Lack of CMMRD-related family history is also typical of previously described case reports [Citation5].
The studied family case confirmed earlier published data that the current clinical criteria for Lynch syndrome are not sensitive enough to identify MSH6 mutation carriers [Citation6]. The c.3514dupA mutation has been previously detected in Lynch syndrome families; however, in the pedigree described here only one CRC apart from that found in the proband was diagnosed.
Teenage colorectal polyposis is usually not sporadic. It can be related to different hereditary disorders: FAP, MUTYH-associated polyposis, juvenile polyposis, Lynch syndrome and CMMRD. Therefore, multi-gene testing by next-generation sequencing panels would be effective in diagnosing all these major genetic disorders. These tests simultaneously analyze a set of the genes that are associated with the polyposis phenotype.
All childhood and teenage malignancy cases (especially hematological, brain and intestinal tract tumors) with café au lait spots should be suspected to be CMMRD. In the studied family the proband’s skin phenotype was caused by biallelic MSH6 gene mutations. Incidentally, her brother had true neurofibromatosis type 1 with de novo mutation in the NF1 gene c.3826C > T (p.Arg1276*). Therefore, we conclude that CMMRD should be suspected in pediatric malignancy cases even if proven neurofibromatosis is present in family members.
One of the important aspects of hereditary polyposis syndromes is their management. Development of a few adenomas can be controlled by regular endoscopic polypectomies over time. However, a high burden of adenomas with severe symptoms or high-grade dysplasia could necessitate total colectomy or proctocolectomy at an early age. At least 40% of CMMRD carriers develop CRC and the median age at diagnosis is as low as 16 years [Citation4]. Therefore, the timing of prophylactic operations is essential. Our patient underwent a prophylactic proctocolectomy at the age of 17. Moreover, besides a large number of adenomas with high-grade dysplasia, one of them contained an intraepithelial adenocarcinoma (pTis).
We conclude that in the case of all childhood and teenage malignancies or colorectal polyposis with café au lait spots, CMMRD should be suspected. Active management, prophylactic surgery and follow-up may prevent some malignancies in CMMRD patients.
References
- Lynch HT, Snyder CL, Shaw TG, et al. Milestones of Lynch syndrome: 1895-2015. Nat Rev Cancer 2015;15:181–94.
- Watson P, Vasen HFA, Mecklin J-P, et al. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer 2008;123:444–9.
- Vasen HFA, Ghorbanoghli Z, Bordeaut F, et al. Guidelines for surveillance of individuals with constitutional mismatch repair-deficiency proposed by the European Consortium, Care for CMMR-D (C4CMMR-D). J Med Genet 2014;51:283–93.
- Wimmer K, Kratz CP, Vasen HFA, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium, Care for CMMRD (C4CMMRD). J Med Genet 2014;51:355–65.
- Levi Z, Kariv R, Barnes-Kedar I, et al. The gastrointestinal manifestation of constitutional mismatch repair deficiency syndrome: from a single adenoma to polyposis-like phenotype and early onset cancer. Clin Genet 2015;88:474–8.
- Sjursen W, Haukanes BI, Grindedal EM, et al. Current clinical criteria for Lynch syndrome are not sensitive enough to identify MSH6 mutation carriers. J Med Genet 2010;47:579–85.