
Prolymphocytic leukemia (PLL) is a rare, aggressive lymphoid malignancy, characterized by marked lymphocytosis (prolymphocytes >55%) and splenomegaly, accounting for 2% of all mature lymphocytic leukemias. There are two subtypes of PLL, B-cell prolymphocytic leukemia (B-PLL) and T-cell prolymphocytic leukemia (T-PLL). Morphologically, B-PLL is identical to T-PLL, but extramedullary features are less common, with favorable survival (3 years vs. 7 months) [Citation1]. More than half of patients with B-PLL carry abnormalities in TP53 tumor suppressor gene. The most common chromosome abnormality in T-PLL is abnormality of chromosome 14 (80%), and Trisomy 8 (53%) [Citation2]. Treatment of B-PLL is extrapolated from chronic lymphocytic leukemia (CLL), and alemtuzumab-based chemotherapy improved survival of T-PLL patients.
The development of a second malignancy is a serious complication of prolonged survival in cancer patients, which accounts for 16% of incident cancers in the USA [Citation3]. Immunologic defects [Citation4], genetic susceptibility [Citation5] and treatment-related factors [Citation6] increase risk of second cancers in CLL, however no reports addressed this issue in PLL. Therefore, I evaluated the overall risk of second primary malignancies (SPMs) after six months diagnosis of 339 patients with a first primary PLL who were selected from the Surveillance, Epidemiology, and End Results (SEER) 13 registries (1992–2012) of US National Cancer Institute (www.seer.cancer.gov). We excluded cases diagnosed by autopsy or reported by death certificate. The standardized incidence ratio (SIR) with corresponding 95% confidence interval (95% CI) was calculated by SEER*stat software (version 8.2.1).
Of the total 399 PLL patients, the median age at diagnosis was 69 years (range 11–100 years), 60% male (male-female ratio of 1.5:1), and 82.6% white (). There were 94 cases (27.7%) with T-PLL, 71 cases with B-PLL (20.9%), and 174 cases (51.4%) with PLL-not otherwise specified (PLL-NOS). The median latency time to develop 29 SPMs among 399 PLL patients was 41 months (range 8–174 months). As shown in a , the overall risk of SPMs was relatively constant between latency period 1–4 years (SIR 1.52) and ≥5 years (SIR 1.77).
Table 1. The incidence rates of second malignancies in patients with prolymphocytic leukemia (PLL) categorized by clinical features and latency interval, USA, 1992–2012.
With a median follow-up of 26 months (range 6–244 months) and contributed 1119 person-years (PY), 26 patients (6.5%) developed 29 SPMs during the study period. The most common second cancers was non-Hodgkin lymphoma (NHL) (n = 8), multiple myeloma (MM) (n = 4), digestive system cancers (n = 3), leukemias (n = 2), urinary system cancers (n = 2), and prostate cancers (n = 2) (). The solid cancers have been reported in 3.8% (n = 13) of patient cohort, compared to 6% that reported in CLL patients received fludarabine, cyclophosphamide and rituximab [Citation6]. We also observed eight of 339 patients (2.3%) developed NHL, however misclassification of PLL is inevitable [Citation7]. Of note, Richter syndrome occurs in approximately 2–10% of CLL patients [Citation8].
Figure 1. Second malignancies in patients with prolymphocytic leukemia compared with general population for different tumor types, SEER, 1992–2012.
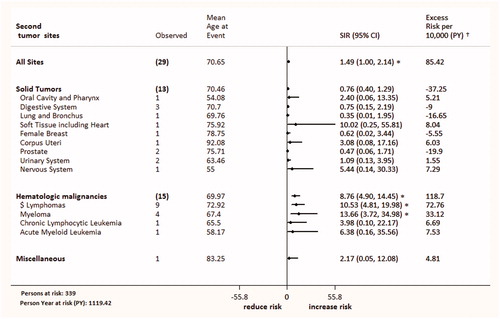
Regarding CLL patients, the risk of second cancers is twice higher than general population in a recent reports [Citation6,Citation9]. We found that the overall risk of all new malignancies in PLL patients was 49% higher than the malignancy in the general populations [Observed (O) 29; SIR 1.49; 95% CI 1–2.14], and an absolute excess risk (AER) of 85.42 per 10 000 PY. It seems that risk of a SPMs is significantly higher in T-PLL (SIR 2.92), compared with B-PLL (SIR 1.93) and PLL-NOS (SIR 1.05).
Compared with the US general population, the overall risk for hematologic malignancies in PLL patients was significantly increased by 7.92-fold. However, the overall risk of a SPMs was non-significantly lower for solid cancers (SIR 0.76; 95% CI 0.4–1.29). This pattern of cancer excesses is in line with a second cancers reported in CLL [Citation9]. The risk of hematological malignancies was remarkably increased for lymphomas (SIR 10.53) and MM (SIR 13.66) (). The most frequent lymphomas was NHL (n = 8), with risk of 9.86-fold. In one report [Citation10], the risk of NHL after CLL was 2.73-fold higher than expected. In a SEER database study [Citation11], the risk of MM after CLL/small lymphocytic lymphoma (SLL) diagnosis was non-significantly increased (SIR 1.13; 95% CI 0.69–1.75).
We observed non-significantly higher risk of a SPMs in females than males (SIR 1.79 vs. 1.36). In literature, whether female patients with CLL are more prone to develop second cancers is a matter of debate [Citation6,Citation9]. Regarding the age groups, we found 3.27-fold excess risk of a SPMs among young patients (<60 years) compared to 1.25-fold in older patients (≥70 years) (). Of note, young patients with PLL survive longer [Citation12,Citation13], and more likely to receive an aggressive therapy. In contrast, two reports from MD Anderson Cancer Center reported that patients aged >60 years have a higher tendency for second cancers, whether in treated [Citation6] or untreated CLL patients [Citation9]. In the current work, the risk of a SPM is remarkably increased only in the recent calendar period; 2007–2012 (SIR 3.64; 95% CI 1.66–6.91; AER 374.32) (). Introduction of aggressive therapy and early surveillance could in part explain increased risk of a SPMs in a recent times.
Several hypotheses suggesting an increased risk of second malignancies in T-PLL patients of the current work: (1) T-PLL is genetically unstable [Citation14], with chromosomal aberrations might occur during the course of the disease [Citation15], which could increase the risk of a second cancers. (2) T-PLL is an extramedullary diseases [Citation16], as malignant T cell can infiltrate organs/tissues (liver, spleen, lymph node, skin, central nervous system, pleura and peritoneum). T-PLL patients with extramedullary diseases had aggressive course with chemoresistance [Citation1]. (3) Although autoimmune disorders are rarely reported in PLL cases [Citation17,Citation18], it may be play a role in a subsequent cancers. (4) Treatment-related immune dysfunction is another possible risk factor. Noticeable, alemtuzumab is the initial treatment of choice in T-PLL, but did not increase the risk of second malignancies following kidney transplantation [Citation19]. In the setting of CLL, alemtuzumab did not increase the incidence of Richter’s transformation [Citation20]. (5) Similar to CLL [Citation21], immunodeficiency of PLL may predispose to the increased risk of a second cancers. (6) Finally, misclassification/diagnosis of PLL is inevitable as there was considerable overlap between the features of B-PLL and mantle cell lymphoma [Citation7,Citation22], hairy cell leukemia and splenic B-cell lymphomas [Citation23].
In conclusion, the overall risk of second malignancies in PLL patients was 49% higher than expected. The risks were significantly increased, for NHL (SIR 9.86) and MM (SIR 13.66). However, the risk of SPMs was non-significantly lower for solid cancers (SIR 0.76).
Funding
This work is not funded.
Disclosure statement
The author declares no conflicts of interests.
References
- Dearden C. How I treat prolymphocytic leukemia. Blood. 2012;120:538–551.
- Matutes E, Brito-Babapulle V, Swansbury J, et al. Clinical and laboratory features of 78 cases of T-prolymphocytic leukemia. Blood. 1991;78:3269–3274.
- Travis LB, Rabkin CS, Brown LM, et al. Cancer survivorship–genetic susceptibility and second primary cancers: research strategies and recommendations. J Natl Cancer Inst. 2006;98:15–25.
- Hisada M, Biggar RJ, Greene MH, et al. Solid tumors after chronic lymphocytic leukemia. Blood. 2001;98:1979–1981.
- Goldin LR, Pfeiffer RM, Li X, et al. Familial risk of lymphoproliferative tumors in families of patients with chronic lymphocytic leukemia: results from the Swedish Family-Cancer Database. Blood. 2004;104:1850–1854.
- Benjamini O, Jain P, Trinh L, et al. Second cancers in patients with chronic lymphocytic leukemia who received frontline fludarabine, cyclophosphamide and rituximab therapy: distribution and clinical outcomes. Leuk Lymphoma. 2015;56:1643–1650.
- van der Velden VH, Hoogeveen PG, de Ridder D, et al. B-cell prolymphocytic leukemia: a specific subgroup of mantle cell lymphoma. Blood. 2014;124:412–419.
- Parikh SA, Kay NE, Shanafelt TD. How we treat Richter syndrome. Blood. 2014;123:1647–1657.
- Tsimberidou AM, Wen S, McLaughlin P, et al. Other malignancies in chronic lymphocytic leukemia/small lymphocytic lymphoma. J Clin Oncol. 2009;27:904–910.
- Schöllkopf C, Rosendahl D, Rostgaard K, et al. Risk of second cancer after chronic lymphocytic leukemia. Int J Cancer. 2007;121:151–156.
- Morton LM, Curtis RE, Linet MS, et al. Second malignancy risks after non-Hodgkin’s lymphoma and chronic lymphocytic leukemia: differences bylymphoma subtype. J Clin Oncol. 2010;28:4935–4944.
- Herling M, Patel KA, Teitell MA, et al. High TCL1 expression and intact T-cell receptor signaling define a hyperproliferative subset of T-cellprolymphocytic leukemia. Blood. 2008;11:328–337.
- Hercher C, Robain M, Davi F, et al. A multicentric study of 41 cases of B-prolymphocytic leukemia: two evolutive forms. Leuk Lymphoma. 2001;42:981–987.
- Maljaei SH, Brito-Babapulle V, Hiorns LR, et al. Abnormalities of chromosomes 8, 11, 14, and X in T-prolymphocytic leukemia studied by fluorescence in situ hybridization. Cancer Genet Cytogenet. 1998;103:110–116.
- Soulier J, Pierron G, Vecchione D, et al. A complex pattern of recurrent chromosomal losses and gains in T-cell prolymphocytic leukemia. Genes Chromosomes Cancer. 2001;31:248–254.
- Valbuena JR, Herling M, Admirand JH, et al. T-cell prolymphocytic leukemia involving extramedullary sites. Am J Clin Pathol. 2005; 123:456–464.
- Michallet AS, Lesca G, Radford-Weiss I, et al. T-cell prolymphocytic leukemia with autoimmune manifestations in Nijmegen breakage syndrome. Ann Hematol. 2003;82:515–517.
- Iioka F, Akasaka T, Hayashida M, et al. B-cell prolymphocytic leukemia carrying t(8;14)(q24;q32), associated with both autoimmune hemolytic anemia and pure red cell aplasia. J Clin Exp Hematop. 2014;54:219–224.
- Puttarajappa C, Yabes J, Bei L, et al. Cancer risk with alemtuzumab following kidney transplantation. Clin Transplant. 2013;27:E264–E271.
- Karlsson C, Norin S, Kimby E, et al. Alemtuzumab as first-line therapy for B-cell chronic lymphocytic leukemia: long-term follow-up of clinical effects, infectious complications and risk of Richter transformation. Leukemia. 2006;20:2204–2207.
- Greene MH, Hoover RN, Fraumeni JF Jr, Subsequent cancer in patients with chronic lymphocytic leukemia–a possible immunologic mechanism. J Natl Cancer Inst. 1978;61:337–340.
- Ruchlemer R, Parry-Jones N, Brito-Babapulle V, et al. B-prolymphocytic leukaemia with t(11;14) revisited: a splenomegalic form of mantle cell lymphoma evolving with leukaemia. Br J Haematol. 2004;125:330–336.
- Hoehn D, Miranda RN, Kanagal-Shamanna R, et al. Splenic B-cell lymphomas with more than 55% prolymphocytes in blood: evidence for prolymphocytoid transformation. Hum Pathol. 2012;43:1828–1838.