
The pioneers who discovered the mechanisms for activation of cytotoxic T-cells and developed antibodies which inactivate the T-cell brakes, professor James P Allison, USA, and professor Tasuku Honjo, Japan, have recently been awarded the Nobel Prize in Physiology or Medicine 2018 by the Nobel Assembly at Karolinska Institutet, Stockholm, Sweden. Immunotherapy, particularly the use of immune checkpoint blockers (ICBs) has been approved for standard clinical use in some settings, but not all tumors respond to the treatment. The aim of the present editorial is, therefore, to present the background, mechanisms of action and the current role of ICBs for clinical anticancer therapy.
Immunotherapy has a more than 100 year long history of preclinical and clinical trials. The general success of vaccines to prevent viral and bacterial diseases has inspired the use of vaccines against malignant diseases [Citation1]. Despite many different approaches, the use of vaccines towards cancer cells has largely been of modest clinical benefit [Citation2]. However, modern sequencing technologies have disclosed that cancer cells have many genetic or epigenetic changes that ultimately produce proteins and peptides classified as unique driver mutations or passenger mutations [Citation3]. Despite the great success of imatinib and newer tyrosine kinase inhibitors against the Bcr/Abl oncoprotein in chronic myelogenous leukemia [Citation4], and the success of antagonists of the Her2/neu tyrosin kinase for breast cancer [Citation5], the clinical results of personalized targeted therapy have long lasting effects in only a limited number of patients with systemic disease, and the responses have generally been short lived.
Cancer development is likely associated with immune suppression: cancer develops in patients with HIV infection or patients on immune suppressive therapy after organ transplantations [Citation6,Citation7]. Newer studies have disclosed that the immune system may recognize cancer cells, but also that cancer can avoid immunologic suppression and control. Some facts necessary to understand the clinical use of ICB drugs which are the recent candidates for immunotherapy will be presented.
Mechanisms for activation and inhibition of cytotoxic T-cells
When cancer cells are exposed to chemotherapy, they generally die by apoptosis [Citation8]. Apoptosis was previously considered not to stimulate an immunological response, in contrast to necrosis which typically elicits an inflammatory response [Citation9]. These two separate mechanisms of dying are in fact interrelated and autophagy seems to be involved in selection between apoptosis and necrosis [Citation10]. Both mechanisms expose tumor-related antigens (neoantigens) which the immune system can identify and direct both the innate immune system and cytotoxic T-lymphocytes (CD8 T-cells) against [Citation11–13]. A clear relation between the number of neoantigens in different tumor entities and their response to immune stimulating therapy by ICBs has been demonstrated [Citation14–16]. Tumors with high number of mutations exhibiting microsatellite instability (MSI) seem especially to be good candidates for ICBs [Citation17,Citation18]. Genetic mutation analyses revealed MSI in 3.8% of tumor and normal tissues assessed from 39 cancer types [Citation19]. However, only a minority of the neoantigens produced by DNA mutations seems to be recognized by CD8 T-cells [Citation20].
To understand the mechanism of action of ICBs, some basic insight into the activation and function of CD8 T-cells are necessary. Cells dying after cancer therapy may undergo a process termed immunogenic cancer cell death [Citation21,Citation22]. In addition to neoantigens, tumor cells exposed to stress including therapeutic methods (radiation therapy, chemotherapy, and hyperthermia) also transfer internal molecules to the surface of the tumor cells as danger or damage associated molecular patterns (DAMPs) which function as semaphores for the immune system’s immature antigen presenting cells (APC) [Citation23–25]. Typical DAMPs are calreticulin, high mobility group box 1 (HMGB1), heat shock protein 70 and 90 (Hsp 70, Hsp 90), extracellular adenosine triphosphate (ATP), S100A4, and uric acid [Citation26,Citation27]. The chemokine axis CXCR4–CXCL12 and DAMPs act also as chemo-attractants which activate dendritic cells and attracts macrophages, granulocytes, and eosinophils [Citation28–31]. The innate immune system is also involved in anticancer immunity by stimulation of dendritic cells and by natural killer cells responding to DAMP’s [Citation32].
The tumor antigens include different overexpressed antigens, cancer testis antigens, and mutated tumor neoantigens [Citation33]. It seems that chaperoning proteins like Hsp 70, Hsp 90, and Hsp 27 bind to antigen peptides which are then presented for immature APC [Citation34]. The peptide antigens from inside of the tumor cells then bind to the major histocompatibility complex type I (MHC I) on APC [Citation35,Citation36]. The human homologs of MHC I and II are termed human leukocyte antigen (HLA) complex [Citation35]. When the antigens are bound to MHC I on APCs these cells migrate to regional lymph nodes where they mature to dendritic cells which present the antigens bound to MHC I to a specific receptor, T cell receptor (TCR) on CD8 T-cells or macrophages [Citation36,Citation37]. The discovery of these mechanisms was pioneered by Allison and his colleagues among others, who disclosed that in order to activate the CD8 T-cell a second signal must be simultaneously sent by binding of the B7 receptor on the APC/dendritic cell to the CD28 ligand on the CD8 T-cell [Citation38]. Later it was shown that a molecular homolog of CD28 termed cytotoxic T-lymphocyte antigen 4 (CTLA-4) bound even stronger to the B7 receptors (CD80 and CD86) [Citation39,Citation40]. CTLA-4 is mainly expressed on CD4 and CD8 T-cells [Citation41]. A seminal study in mice then documented that CTLA-4 inhibited initial priming and activation of CD8 T-lymphocytes [Citation42]. Antibodies against CTLA-4 can also deplete immunosuppressive CD4 T-cells (regulatory T-cells) [Citation43,Citation44]. The recent achievements in immunotherapy are largely based on further insight on the molecules and their receptors which normally control immunological responses as needed: activating immune responses or damping the immune effector cells when an infectious agent is controlled or to avoid autoimmune conditions (). The level of antigens and the degree of naïve CD8 T-cells in the draining lymph nodes seems to determine whether the immune system responds with tolerance or an active anticancer response, both processes may take place simultaneously changing the immunogenicity of a tumor, a process now termed cancer immunoediting [Citation45,Citation46].
Figure 1. Review of major mechanisms for activation of CD-8 T-cells by neoantigens from a tumor. First immature antigen presenting cells (APC) is activated to mature dendritic cells (DC) by binding of neoantigens to major histocompatibility complex type I (MHC I). Co-stimulation for activation of CD8 T-cells is illustrated by the binding of CD28 receptors on T-cells to either B7-1 or B7-2 ligands on the mature DC. The inhibiting receptor CTLA-4 is also illustrated. The main inhibition of peripheral effect of T-cell is exerted by binding of programed death receptor 1 (PD-1) to either programed death ligand 1 or 2 (PD-L1 or PD-L2). Immune checkpoint blockers (ICBs) are represented with antibodies raised against the blocking molecules (marked in red). Interferons (IFN) together with transforming growth factor β (TGFβ) protect tumor cells from immunologic inactivation. Stimulating signals marked (+), inhibition marked (–) (illustration: Kristin Risa).
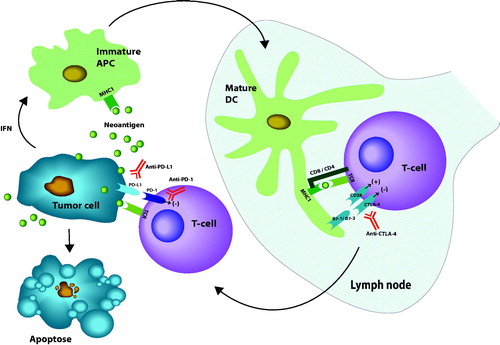
When studying programed cell death in a murine hybridoma and hematopoietic progenitor cell line, Honjo and his group identified a gene they termed programed death 1 gene (PD-1 gene) coding for a surface receptor of the Ig superfamily [Citation47,Citation48]. They then discovered a ligand (PD-L1) as a member of the B7 family on APCs that dampen the CD8 T-cell response [Citation49]. Later, it was shown that cancer cells express programed death 1 or 2 ligands (PD-L1, PD-L2) on the surface which bind to programed death 1 (PD-1) receptors on CD8 T-cells and thereby exploit the natural immune damping system for inactivation of CD8 T-cells, which is expressed on macrophages and bone marrow derived cells [Citation50–53]. By reducing this mechanism antibodies against PD-1 or PD-L1 can re-invigourate exhausted CD8 T-cells to attack tumor cells [Citation54].
Clinical use of immune checkpoint inhibitors
Antibodies inhibiting CTLA-4 (ipilimumab, tremelimumab) and PD-1 (pembrolizumab, nivolumab) and PD-L1 (durvalumab, avelumab, atezolizumab) are approved by U.S. Food and Drug Administration (FDA) or European Medicines Agency (EMA) for clinical use (). The clinical indications are given in ; therefore, only some typical studies will briefly be presented here.
Table 1. Clinical indications approved by U.S. Food and Drug Administration (FDA) and/or European Medicines Agency (EMA) for immune checkpoint blockers. Only the year of first approval for an indication of a drug given in the table.
The initial dose finding studies with ipilimumab in advanced malignant melanoma reported only objective responses in 10% of the patients [Citation55]. For pembrolizumab the response was better (20–30%), and some long duration responses were observed. The two drugs were then combined, based on the rationale that CTLA-4 inhibition with ipilimumab stimulates CD8 T-cells locally, while PD-L1 inhibition with pembrolizumab or nivolumab works on exhausted CD8 T-cells at any site. The combination yielded a response rate of 50–70% in advanced malignant melanoma and 3–5 year survival of 60% is now reported [Citation56,Citation57]. In a randomized study using ipilimumab versus placebo as adjuvant therapy after resection of malignant melanoma stage III, the survival was 41% and 30%, respectively [Citation58]. In two randomized studies including resected malignant melanoma stage III or IV comparing pembrolizumab versus placebo and nivolumab versus ipilimumab, early results show best durable effect of the of pembrolizumab and nivolumab arms compared to the control groups with placebo and ipilimumab, respectively (difference about 10% at 5 and 2 years) [Citation59,Citation60]. Grades 3–4 toxicity was 14% for nivulomab versus 46% for ipilimumab.
In advanced non-small cell lung cancer (NSCLC), the objective response rate was 19%, after treatment with pembrolizumab [Citation61]. Treatment with nivolumab and pembrolizumab improved median progression free survival in NSCLC with 3–4 months more than docetaxel and platinum-based regimens [Citation62–64]. Combining nivolumab and ipilimumab in different schedules for advanced NSCLC yielded confirmed partial responses in 47% and 38% in the best combinations, and median follow up was not yet achieved after 12 months follow up [Citation65].
Pembrolizumab monotherapy yielded 18% objective responses in recurrent or metastatic head and neck squamous cell carcinomas patients [Citation66]. Median overall survival was 13 months. In a randomized trial using nivolumab versus standard therapy for recurrent or metastatic head and neck cancer, 1-year survival was 36% and 17%, respectively [Citation67]. In Merkel cell carcinoma, a rare aggressive skin cancer (related to a polyoma virus in 80%), objective response was observed in 56% of 28 patients treated with pembrolizumab as first line therapy and 33% in 88 chemotherapy refractory patients treated with avelumab [Citation68,Citation69]. Some patients obtained long duration of response.
Renal cell cancer is generally resistant to chemotherapy, but metastatic disease is currently treated by targeted agents like temsirolimus and sorafenib. When nivolumab was compared with everolimus in metastatic renal cell cancer 25% and 5% obtained objective responses, respectively, but few obtained a complete response [Citation70]. In advanced renal cell cancer patients the combination of nivolumab and ipilimumab yielded 75% survival at 18 months in contrast to 60% for sunitinib [Citation71]. Several clinical studies are now ongoing testing ICBs in renal cell cancer patients [Citation72].
In early bladder cancer, immunotherapy with BCG-instillations into the bladder is standard therapy. In a series of trials different ICBs (CTLA-4 blockers ipilimumab, tremelimumab, PD-1blockers nivolumab, and pembrolizumab or PD-L1 blockers atezolizumab, avelumab, durvalumab) have yielded objective responses of 13–30% in advanced urothelial tumors, but generally short lived [Citation73,Citation74]. Only pembrolizumab increased significantly median overall survival from 7.4 months in controls to 10.3 months [Citation75].
Several types of immunotherapy have been tested in gastrointestinal cancers [Citation76]. In therapy resistant PD-L1 positive esophageal cancer an objective response rate of 30% were seen in 23 patients which lasted for 15 months [Citation77]. In a phase II study using nivolumab and ipilimumab in MSI high colorectal cancer, objective response was seen in 55% of 119 patients, and progression free and overall survival were 71% and 85%, respectively [Citation78]. Eight had responses lasting at least 12 months. In unresectable metastatic anal cancer, treatment with nivolumab resulted in 24% objective responses, and the median progression free survival was 4.1 months [Citation79].
Very promising results were published in Hodgkin’s lymphoma patients treated with nivolumab or pembrolizumab where objective response was observed in 65–87%, of these 16–20% were long lasting complete responses [Citation80–82]. Interestingly, Reed Sternberg cells in Hodgkin’s lymphoma express PD-L1 to secure immunological protection. In other types of lymphomas objective responses ranging from 20 to 40% have been reported [Citation83,Citation84].
Biomarkers
Tumors may express PD-L1 or PD-L2 on the surface membrane to protect them from cytotoxic CD8 T-cells. Some studies have reported higher response rate in tumors which express PD-L1 or PD-L2 on the membrane [Citation85,Citation86], but the level of expression varies from tumor to tumor, and responses are also seen in tumors lacking these markers. The density of infiltrating CD8 T-cells in tumors and number of neoantigens in tumor cells are also linked with better tumor response [Citation86,Citation87]. Immune gene signatures and markers in serum are under investigation as biomarkers for response to ICB therapy, but we still lack reliable markers to select patients suitable for ICBs. Documented MSI high or mismatch repair deficiency in any tumor is now approved by FDA as an indication for use of ICBs.
Resistance
It should be noted that an increase in tumor volume may occur for some time after initiation of ICB therapy before tumor regression, probably due to infiltration of cytotoxic CD8 T-cells or edema [Citation88]. Classical RECIST v 1.1 criteria for response evaluation may underestimate the benefit of pembrolizumab in approximately 15% of melanoma patients [Citation89]; therefore, the criteria for response has been specified to immune-related RECIST and immune RECIST criteria [Citation90].
Resistance to ICB treatment can be caused by primary properties in the tumor or the host before treatment or as acquired as a response to the treatment, see . Examples of adaptive tumor responses are reduction of tumor neoantigens or changes in the antigen presentation like mutations in β-2 microglobulin as part of the HLA complex or reduced maturation of the dendritic cells by changes in DAMPs [Citation91–93].
Table 2. Mechanisms for primary and acquired resistance to immune checkpoint inhibitors.
The structure and adaptive changes in the microenvironment like tumor-associated macrophages type 2, cancer associated fibroblasts and presence of proangiogenic growth factors, infiltration of regulatory T-cells (T-reg), myeloid derived cells or cytokines (CXCR4 –CXCL12 axis) all influence the contact between the CD8 T-cells and tumor cells [Citation94,Citation95]. Other mechanisms may be exhaustion of CD8 T-cells or upregulation of other inhibitory immune checkpoints like TIM-3, LAG-3, TIGIT, or VISTA [Citation96–99]. Some of these checkpoints are targets for clinical trials. The host gut microbiome is important for general priming of the immune system to discern between own cells and infectious agents by influencing the microenvironment [Citation100,Citation101]. Commensal bacterial flora is also associated with the effect of ICBs targeting the PD-1 axis [Citation102–106]. Use of antibiotics during ICB treatment has been linked to reduced clinical benefit [Citation107].
ICB treatment may also in some patients cause an accelerated proliferation of tumor cells termed hyperprogression [Citation108–110]. The rapid clinical deteroriation within two months seems linked to amplifications of MDM2 and MDM4 and aberrations in the EGFR genes [Citation111].
Toxicity
Immunotherapy by ICBs is well tolerated by most patients, with treatment-related grade 3–4 adverse events in 16% after nivolumab and 27% after ipilimumab, but combination of these cause serious side effects in 55% [Citation112]. However, side effects like endocrinological adverse effects (hypophysitis, thyroiditis), cutaneous side effects, gastrointestinal side effects (colitis), cardiac and pulmonary toxicities are largely manageable [Citation113–115]. Autoimunity may therefore limit the use of ICB treatment in some patients [Citation116]. The effect of antibodies blocking CTLA-4 on CD4 regulatory T-cells may explain the higher rate of autoimmune toxicity related to this class of ICBs [Citation44]. Fortunately, the response rate and duration of response remained high in malignant melanoma patients who had to stop combined treatment with nivolumab and ipilimumab due to side effects [Citation117].
Prospects
It is encouraging to see clinical responses in tumors generally not expected to respond or have a low response rate to available therapy. However, one must acknowledge that many patients does not respond to ICB monotherapy as the objective response rates are generally 20–30%, with substantial higher rates for malignant melanoma and Hodgkin’s lymphoma [Citation118]. Whereas most responses are short lived, the combination of antibodies targeting CTLA-4 and PD-1/PD-L1/PD-L2 seems to yield more durable responses, but the side effects also increase.
Combinations of ICB therapy with targeted drugs [Citation119–121], chemotherapy [Citation122,Citation123] and radiation [Citation124,Citation125] are currently in clinical trials. It is important that CD8 T-cells may attack cancer cells in any site in the body, thus being a systemic anticancer agent. Experience indicates that most anticancer drugs as well as immunotherapy are most effective when the cancer cell burden is limited.
As mentioned above, ICBs are effective as adjuvant therapy after surgery for melanoma patients [Citation58,Citation60,Citation126]. Both experimental studies [Citation127] and recent clinical experience in resectable lung cancer [Citation128], malignant melanoma [Citation129,Citation130], urothelial bladder cancer [Citation131], and MSI high colon cancer [Citation132] show that ICB can totally eradicate a tumor after one to three courses when administered as neoadjuvant therapy before surgery. More than 60 clinical trials are ongoing using neoadjuvant ICB therapy [Citation133].
Immune checkpoint inhibition is now a part of our standard armamentarium to control malignant diseases. As there are many more signal molecules steering the activity of the immune responding cells, of which several are under active investigation, the role of this class of drugs are promising. The expectation of this novel group of anticancer agents is high, but ICBs final role will have to be documented in clinical trials. A major limitation for general use in palliative settings is the high cost of ICBs, especially when combination of several substances is used. However, if the promising neoadjuvant data can be reproduced, a substantially reduced cost will follow opening for a wider use of these drugs.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Antonia S, Mule JJ, Weber JS. Current developments of immunotherapy in the clinic. Curr Opin Immunol. 2004;16:130–136.
- Klebanoff CA, Acquavella N, Yu Z, et al. Therapeutic cancer vaccines: are we there yet? Immunol Rev. 2011;239:27–44.
- McGranahan N, Favero F, de Bruin EC, et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med. 2015;7:283ra254.
- Awan FT, Byrd JC. New strategies in chronic lymphocytic leukemia: shifting treatment paradigms. Clin Cancer Res. 2014;20:5869–5874.
- Baselga J, Coleman RE, Cortes J, et al. Advances in the management of HER2-positive early breast cancer. Crit Rev Oncol Hematol. 2017;119:113–122.
- Grulich AE, van Leeuwen MT, Falster MO, et al. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet. 2007;370:59–67.
- Yanik EL, Clarke CA, Snyder JJ, et al. Variation in cancer incidence among patients with ESRD during kidney function and nonfunction intervals. J Am Soc Nephrol. 2016;27:1495–1504.
- Savill J, Dransfield I, Gregory C, et al. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975.
- Green DR, Oguin TH, Martinez J. The clearance of dying cells: table for two. Cell Death Differ. 2016;23:915–926.
- Goodall ML, Fitzwalter BE, Zahedi S, et al. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell. 2016;37:337–349.
- Branca MA. Rekindling cancer vaccines. Nat Biotechnol. 2016;34:1019–1024.
- Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581.
- Heemskerk B, Kvistborg P, Schumacher TN. The cancer antigenome. EMBO J. 2013;32:194–203.
- Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421.
- Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128.
- Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med. 2017;377:2500–2501.
- Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413.
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015; 348:69–74.
- Bonneville R, Krook MA, Miya J, et al. Landscape of microsatellite instability across 39 cancer types. Precis Oncol. 2017;0:1–15.
- Stronen E, Toebes M, Kelderman S, et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016;352:1337–1341.
- Galluzzi L, Buque A, Kepp O, et al. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111.
- Tesniere A, Panaretakis T, Kepp O, et al. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2008;15:3–12.
- Fuchs EJ, Matzinger P. Is cancer dangerous to the immune system? Semin Immunol. 1996;8:271–280.
- Garg AD, Galluzzi L, Apetoh L, et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol. 2015;6:e588.
- Pradeu T, Cooper EL. The danger theory: 20 years later. Front Immunol. 2012;3:287.
- Eisenbacher JL, Schrezenmeier H, Jahrsdorfer B, et al. S100A4 and uric acid promote mesenchymal stromal cell induction of IL-10+/IDO + lymphocytes. J Immunol. 2014;192:6102–6110.
- Sharabi AB, Lim M, DeWeese TL, et al. Radiation and checkpoint blockade immunotherapy: radiosensitisation and potential mechanisms of synergy. Lancet Oncol. 2015;16:e498–e509.
- Lotfi R, Kaltenmeier C, Lotze MT, et al. Until death do us part: necrosis and oxidation promote the tumor microenvironment. Transfus Med Hemother. 2016;43:120–132.
- Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80.
- Ohta A. A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol. 2016;7:e109.
- Vega VL, Rodriguez-Silva M, Frey T, et al. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J Immunol. 2008;180:4299–4307.
- Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol. 2015;33:445–474.
- Esposito A, Criscitiello C, Curigliano G. Immune checkpoint inhibitors with radiotherapy and locoregional treatment: synergism and potential clinical implications. Curr Opin Oncol. 2015;27:445–451.
- Wells AD, Malkovsky M. Heat shock proteins, tumor immunogenicity and antigen presentation: an integrated view. Immunol Today. 2000;21:129–132.
- Horton R, Wilming L, Rand V, et al. Gene map of the extended human MHC. Nat Rev Genet. 2004;5:889–899.
- Rock KL, Reits E, Neefjes J. Present yourself! By MHC class I and MHC class II molecules. Trends Immunol. 2016;37:724–737.
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61.
- Harding FA, McArthur JG, Gross JA, et al. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature. 1992;356:607–609.
- Allison JP, Krummel MF. The Yin and Yang of T cell costimulation. Science. 1995;270:932–933.
- Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465.
- McCoy KD, Le Gros G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol Cell Biol. 1999;77:1–10.
- Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413.
- Curran MA, Montalvo W, Yagita H, et al. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–4280.
- Simpson TR, Li F, Montalvo-Ortiz W, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210:1695–1710.
- Dunn GP, Bruce AT, Ikeda H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998.
- Dunn GP, Ikeda H, Bruce AT, et al. Interferon-gamma and cancer immunoediting. Immunol Res. 2005;32:231–245.
- Ishida Y, Agata Y, Shibahara K, et al. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887–3895.
- Iwai Y, Ishida M, Tanaka Y, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293–12297.
- Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034.
- Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375:1767–1778.
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264.
- Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214.
- Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461.
- Iwai Y, Hamanishi J, Chamoto K, et al. Cancer immunotherapies targeting the PD-1 signaling pathway. J Biomed Sci. 2017;24:26.
- Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155–164.
- Callahan MK, Kluger H, Postow MA, et al. Nivolumab plus ipilimumab in patients with advanced melanoma: updated survival, response, and safety data in a phase I dose-escalation study. JCO. 2018;36:391–398.
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345–1356.
- Eggermont AM, Chiarion-Sileni V, Grob JJ, et al. Prolonged survival in stage III melanoma with ipilimumab adjuvant therapy. N Engl J Med. 2016;375:1845–1855.
- Eggermont AMM, Blank CU, Mandala M, et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N Engl J Med. 2018;378:1789–1801.
- Weber J, Mandala M, Del Vecchio M, et al. Adjuvant nivolumab versus ipilimumab in resected stage III or IV melanoma. N Engl J Med. 2017;377:1824–1835.
- Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–2028.
- Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–1639.
- Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–1550.
- Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823–1833.
- Hellmann MD, Rizvi NA, Goldman JW, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017;18:31–41.
- Chow LQM, Haddad R, Gupta S, et al. Antitumor activity of pembrolizumab in biomarker-unselected patients with recurrent and/or metastatic head and neck squamous cell carcinoma: results from the phase Ib KEYNOTE-012 expansion cohort. J Clin Oncol. 2016;34:3838–3845.
- Ferris RL, Blumenschein G Jr, Fayette J, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856–1867.
- Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17:1374–1385.
- Nghiem PT, Bhatia S, Lipson EJ, et al. PD-1 blockade with pembrolizumab in advanced Merkel-Cell carcinoma. N Engl J Med. 2016;374:2542–2552.
- Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–1813.
- Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378:1277–1290.
- Ross K, Jones RJ. Immune checkpoint inhibitors in renal cell carcinoma. Clin Sci. 2017;131:2627–2642.
- Giridhar KV, Kohli M. Management of muscle-invasive urothelial cancer and the emerging role of immunotherapy in advanced urothelial cancer. Mayo Clin Proc. 2017;92:1564–1582.
- Kamat AM, Bellmunt J, Galsky MD, et al. Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of bladder carcinoma. J Immunother Cancer. 2017;5:68.
- Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376:1015–1026.
- Wang J, Reiss KA, Khatri R, et al. Immune therapy in GI malignancies: a review. J Clin Oncol. 2015;33:1745–1753.
- Doi T, Piha-Paul SA, Jalal SI, et al. Safety and antitumor activity of the anti-programmed death-1 antibody pembrolizumab in patients with advanced esophageal carcinoma. Jco. 2018;36:61–67.
- Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–1191.
- Morris VK, Salem ME, Nimeiri H, et al. Nivolumab for previously treated unresectable metastatic anal cancer (NCI9673): a multicentre, single-arm, phase 2 study. Lancet Oncol. 2017;18:446–453.
- Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372:311–319.
- Maly J, Alinari L. Pembrolizumab in classical Hodgkin's lymphoma. Eur J Haematol. 2016;97:219–227.
- Villasboas JC, Ansell S. Checkpoint inhibition: programmed cell death 1 and programmed cell death 1 ligand inhibitors in Hodgkin lymphoma. Cancer J. 2016;22:17–22.
- Lesokhin AM, Ansell SM, Armand P, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib study. J Clin Oncol. 2016;34:2698–2704.
- Xu-Monette ZY, Zhou J, Young KH. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood. 2018;131:68–83.
- Chen PL, Roh W, Reuben A, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune check point blockade. Cancer Discov. 2016;6:827–837.
- Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016;17:e542–e551.
- Topalian SL, Taube JM, Anders RA, et al. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–287.
- Chiou VL, Burotto M. Pseudoprogression and immune-related response in solid tumors. J Clin Oncol. 2015;33:3541–3543.
- Hodi FS, Hwu WJ, Kefford R, et al. Evaluation of immune-related response criteria and RECIST v1.1 in patients with advanced melanoma treated with pembrolizumab. Jco. 2016;34:1510–1517.
- Beer L, Hochmair M, Prosch H. Pitfalls in the radiological response assessment of immunotherapy. Memo. 2018;11:138–143.
- Kim JM, Chen DS. Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol. 2016;27:1492–1504.
- Patel SJ, Sanjana NE, Kishton RJ, et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548:537–542.
- Sade-Feldman M, Jiao YJ, Chen JH, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8:1136.
- Buque A, Bloy N, Aranda F, et al. Trial Watch: immunomodulatory monoclonal antibodies for oncological indications. Oncoimmunology. 2015;4:e1008814.
- Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322.
- Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity. 2016;44:989–1004.
- Cogdill AP, Andrews MC, Wargo JA. Hallmarks of response to immune checkpoint blockade. Br J Cancer. 2017;117:1–7.
- Lines JL, Sempere LF, Broughton T, et al. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol Res. 2014;2:510–517.
- Marin-Acevedo JA, Dholaria B, Soyano AE, et al. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. 2018;11:39.
- Goldszmid RS, Dzutsev A, Viaud S, et al. Microbiota modulation of myeloid cells in cancer therapy. Cancer Immunol Res. 2015;3:103–109.
- Iida N, Dzutsev A, Stewart CA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967–970.
- Gopalakrishnan V, Helmink BA, Spencer CN, et al. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell. 2018;33:570–580.
- Gopalakrishnan V, Spencer CN, Nezi L, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103.
- Matson V, Fessler J, Bao R, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104–108.
- Routy B, Le Chatelier E, Derosa L, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–97.
- Sivan A, Corrales L, Hubert N, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084–1089.
- Derosa L, Hellmann MD, Spaziano M, et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann Oncol. 2018;29:1437–1444.
- Champiat S, Dercle L, Ammari S, et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin Cancer Res. 2017;23:1920–1928.
- Saada-Bouzid E, Defaucheux C, Karabajakian A, et al. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol. 2017;28:1605–1611.
- Sznol M, Ferrucci PF, Hogg D, et al. Pooled analysis safety profile of nivolumab and ipilimumab combination therapy in patients with advanced melanoma. Jco. 2017;35:3815–3822.
- Kato S, Goodman A, Walavalkar V, et al. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res. 2017;23:4242–4250.
- Malkhasyan KA, Zakharia Y, Milhem M. Quality-of-life outcomes in patients with advanced melanoma: a review of the literature. Pigment Cell Melanoma Res. 2017;30:511–520.
- Brahmer JR, Lacchetti C, Schneider BJ, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice guideline. Jco. 2018;36:1714–1768.
- Hofmann L, Forschner A, Loquai C, et al. Cutaneous, gastrointestinal, hepatic, endocrine, and renal side-effects of anti-PD-1 therapy. Eur J Cancer. 2016;60:190–209.
- Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139–148.
- June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles' heel of cancer immunotherapy? Nat Med. 2017;23:540–547.
- Schadendorf D, Wolchok JD, Hodi FS, et al. Efficacy and safety outcomes in patients with advanced melanoma who discontinued treatment with nivolumab and ipilimumab because of adverse events: a pooled analysis of randomized phase II and III trials. JCO. 2017;35:3807–3814.
- Smyth MJ, Ngiow SF, Ribas A, et al. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol. 2016;13:143–158.
- Karachaliou N, Gonzalez-Cao M, Sosa A, et al. The combination of checkpoint immunotherapy and targeted therapy in cancer. Ann Transl Med. 2017;5:388.
- Poon E, Mullins S, Watkins A, et al. The MEK inhibitor selumetinib complements CTLA-4 blockade by reprogramming the tumor immune microenvironment. J Immunother Cancer. 2017;5:63.
- Zhang J, Bu X, Wang H, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91–95.
- Ariyan CE, Brady MS, Siegelbaum RH, et al. Robust antitumor responses result from local chemotherapy and CTLA-4 blockade. Cancer Immunol Res. 2018;6:189–200.
- Govindan R, Szczesna A, Ahn MJ, et al. Phase III trial of ipilimumab combined with paclitaxel and carboplatin in advanced squamous non-small-cell lung cancer. J Clin Oncol. 2017;35:3449–3457.
- Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;105:256–265.
- Dahl O, Dale JE, Brydøy M. Rationale for combination of radiation therapy and immune checkpoint blockers to improve cancer treatment. Acta Oncol. 2019;58:xx.
- Eggermont AM. Adjuvant ipilimumab in stage III melanoma: new landscape, new questions. Eur J Cancer. 2016;69:39–42.
- Liu J, Blake SJ, Yong MC, et al. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov. 2016;6:1382–1399.
- Forde PM, Chaft JE, Smith KN, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. 2018;378:1976–1986.
- Amaria RN, Reddy SM, Tawbi HA, et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat Med. 2018;24:1649–1654.
- Blank CU, Rozeman EA, Fanchi LF, et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat Med. 2018;24:1655–1661.
- Necchi A, Anichini A, Raggi D, et al. Pembrolizumab as neoadjuvant therapy before radical cystectomy in patients with muscle-invasive urothelial bladder carcinoma (PURE-01): an open-label, single-arm, phase II study. J Clin Oncol. 2018;JCO1801148.
- Chalabi M, Fanchi LF, van den Berg J, et al. Neoadjuvant ipilimumab plus nivolumab in early stage colon cancer. ESMO Congress. 2018;2018. Abstract LBA 37.
- Keung EZ, Ukponmwan EU, Cogdill AP, et al. The rationale and emerging use of neoadjuvant immune checkpoint blockade for solid malignancies. Ann Surg Oncol. 2018;25:1814–1827.
- Winograd R, Byrne KT, Evans RA et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res. 2015;3:399–411.
- Grasso CS, Giannakis M, Wells DK et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov. 2018;8:730–749.
- Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017;168:707–723.
- Gao J, Shi LZ, Zhao H et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016;167:397–404. e399.
- ] Shukla SA, Bachireddy P, Schilling B et al. Cancer-Germline Antigen Expression Discriminates Clinical Outcome to CTLA-4 Blockade. Cell 2018;173:624–633 .e628.
- Andrews MC, Reuben A, Gopalakrishnan V, Wargo JA. Concepts Collide: Genomic, Immune, and Microbial Influences on the Tumor Microenvironment and Response to Cancer Therapy. Front Immunol. 2018;9:946.
- Fearon DT. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol Res. 2014;2:187–193.
- Gordon SR, Maute RL, Dulken BW et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017;545:495–499.
- Pitt JM, Vetizou M, Daillere R et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016;44:1255–1269.
- Koyama S, Akbay EA, Li YY et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501.