
Abstract
Background: Molecular markers may identify subgroups of patients with clinically distinct behavior and response to treatment. In some gastrointestinal tumors, KRAS has prognostic value and negative predictive value. This is the first prospective study to report the outcome of combination chemotherapy in biliary tract cancer patients with KRAS mutation.
Methods: From 2009 to 2015, 25 patients were included from two Scandinavian centers. Main inclusion criteria were non-resectable biliary tract cancer, ECOG performance status 0–2 and tumor KRAS mutation. A bi-weekly cycle of chemotherapy was administered as gemcitabine 1000 mg/m2 and oxaliplatin 85 mg/m2 day 1, followed by 7 days of oral capecitabine 1000 mg/m2. Response evaluation was done every six treatment and the primary endpoint was the fraction with progression free survival (PFS) at 6 months. The study also included a non-preplanned analysis of circulating tumor specific DNA.
Results: Chemotherapy was given for a median of 5 months (range 0–14) and among 17 patients evaluable for response, best responses were complete response (1), partial response (2), and stable disease (14). Eighteen patients had CT-verified progression, six died between evaluations and one patient is still progression-free. Median PFS was 6.8 months (95% CI 3.1–11.0) and median overall survival (OS) was 11.2 months (95% CI 6.6–14.3). The fraction with PFS at 6 months was 52% (95% CI 31–69%). Exploratory analyses found an improved survival in patients with a low level of plasma DNA.
Conclusion: Pretreatment molecular characterization was feasible in BTC, but the rate of KRAS mutations was low. The study met its primary endpoint with a fraction of PFS at six months of 52%. The effect of combination chemotherapy with gemcitabine, oxaliplatin and capecitabine in this selected population was comparable to results from unselected groups with PFS and OS of 6.8 and 11.2 months, respectively.
ClinicalTrials.gov NCT00779454
Introduction
The incidence of biliary tract cancer (BTC) is low [Citation1] and studies with selection to treatment based on molecular subgroups are thus more difficult to conduct. At the time of design of the present study, there was no standard treatment for patients with non-resectable BTC and no markers to support the clinical decision of which treatment to offer [Citation2]. In colorectal cancer, mutations in Kirstein rat sarcoma viral oncogene homolog (KRAS) have proved to be a negative predictive marker of the effect of treatment with epidermal growth factor receptor inhibitors (EGFR-I) [Citation3,Citation4].
KRAS is a signaling molecule in the Raf–mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway, and this key signaling pathway is often upregulated in cancer. Oncogene activation caused by mutations in KRAS increases cell growth and survival [Citation5].
Based on the biologic rationale and the data from colorectal cancer, we designed a study with upfront molecular characterization of KRAS and selection to treatment on the basis of the specific subtype. The first part of the study which evaluated the effect of chemotherapy combined with panitumumab in patients with wild-type tumors has previously been published [Citation6]. In this second part of the study, we evaluated the effect of triple chemotherapy in patients with KRAS mutations. The purpose of this phase II trial was to test if the fraction of progression free survival (PFS) at six months was superior compared to historic control. Post hoc analyses were undertaken to assess the importance of total plasma DNA and tumor specific plasma DNA. Furthermore, the prognostic value of KRAS was evaluated in all patients undergoing systemic therapy for BTC.
Material and methods
Patients
Adult patients at least 18 years of age were eligible if they had unresectable BTC. Resectability was evaluated by a multidisciplinary team with participation of liver surgeons, radiologists, and oncologists. The diagnosis of BTC required a histologic diagnosis of adenocarcinoma or cytological diagnosis of malignant cells from the bile ducts. Furthermore, imaging should support the origin of a primary lesion in the hilar, intrahepatic or extrahepatic bile ducts or the gall bladder and there should be no sign of another primary tumor. The quantity and quality of tumor material should allow KRAS mutation analysis and a mutation should be present. Other inclusion criteria were evaluable, i.e., not necessarily measurable, disease according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0 [Citation7]; no prior chemotherapy for unresectable disease; ECOG performance status of 0, 1 or 2; adequate hematologic function and hepatic function; bilirubinemia ≤3 x upper normal value; adequate renal function; safe birth control in fertile patients; and oral and written informed consent. Patients were excluded if they had neuropathy above grade 1 or other serious medical disease. Ineligible patients were registered on a screening list holding information only about KRAS mutation status, systemic treatment and date of death.
Ethics, approval, registration and quality assurance
The study was conducted in accordance with the Helsinki Declaration of 1975, as revised in 1983. The study was approved by the Ethics Committee in Southern Denmark, the Danish Data Protection Agency and the Danish Medicines Agency. Prospective trial registration was with ClinicalTrials.gov NCT00779454. Monitoring of adherence to Danish Law and Guideline for Good Clinical Practice (GCP) was conducted by an independent, publicly funded GCP Unit.
Study design and treatment
In the overall study, patients were allocated to treatment according to KRAS status. The part reported here included patients with KRAS mutations and is per se a single arm, open label, phase II trial. The primary objective of the study was to measure the fraction of patients with PFS at six months after initiation of the combination of gemcitabine, oxaliplatin and capecitabine as first line treatment to patients with non-resectable BTC. Secondary objectives were to evaluate the response rate and overall survival (OS). The trial protocol is reported in Supplement 1 (online).
Patients received intravenous gemcitabine 1,000 mg/m2 and oxaliplatin 60 mg/m2 day 1 followed by oral capecitabine 1,000 mg/m2 × 2 daily days 1–7. Treatment was repeated every two weeks with no preplanned minimum or maximum.
Safety and tolerability were evaluated according to the NCI Common Terminology Criteria for Adverse Events, version 3 [Citation8]. Protocol guidelines for management of toxicity corresponded to suggestions in the relevant summary of product characteristics.
Tumor and blood analyses
DNA was purified from tumor material using standard methods. Mutation analysis covered exon 2, codon 12 and 13 of KRAS. Specific mutations included KRAS G12A, G12R, G12D, G12C, G12S, G12V and G13D [Citation9]. As part of the study, a biobank was established holding consecutive blood samples. In these blood samples we analyzed total plasma DNA as the gene copy number of Beta-2-Microglobulin [Citation10]. Tumor specific plasma DNA (ctDNA) was analyzed by digital PCR as the fraction of KRAS mutated DNA relative to total DNA [Citation11]. Briefly, 2 ml of plasma was purified on a QiaSymphony robot (Qiagen, Hilden, Germany) and analyzed by qPCR for B2M. The remaining DNA was concentrated and analyzed in two wells by droplet digital PCR (BioRad, Hercules, CA, USA) for specific KRAS mutations using PrimePCR assays (BioRad).
Assessments and end point
Eligibility assessments included computed tomography (CT) scans of the chest, abdomen and pelvis within four weeks from inclusion and blood tests together with medical history taking and clinical examination within two weeks from inclusion. CT was repeated every six treatments corresponding to about 12 weeks interval depending on any treatment delays. After treatment patients were evaluated clinically and with a CT scan every three months. Tumor progression and tumor responses were determined using RECIST 1.0 [Citation7] by the investigator. PFS was defined as time from inclusion to disease progression or death. Patients were censored at the last CT scan if without progression or death or at registration if there were no tumor assessments after inclusion. The fraction of PFS at six months was calculated as the number of patients alive without progression at the evaluation around six months (± four weeks) divided by the total number of included patients. OS was defined as time from inclusion in the trial to death from any cause. Patients were censored at the date last known alive. Adverse events were evaluated according to CTCAE version 3 [Citation8].
Statistics
The trial is based on a modification of Simon’s two-stage minimax design [Citation12] with six months’ PFS instead of response. The target was set at a fraction of 30% of all included patient alive and without progression six months after inclusion in the study. The treatment would have no interest if fewer than 10% achieve PFS of more than six months. With a significance level of 5% and a power of 80%, the trial should include 15 patients in the first part. If two or more of these patients have a PFS of ≥6 months, the study should include a total of 25 patients. If more than six patients have a PFS of at least six months, the study is considered positive and the treatment regimen is candidate for further clinical testing.
Non-parametric methods are used to establish and compare patient characteristics, toxicity and response rates. PFS and OS are calculated using the Kaplan-Meier method and compared with log-Rank test.
Results
Patients and treatment
From 2009 to 2015, 25 patients were included from two Scandinavian centers. There were more women (n = 16) compared to men (n = 9) and the median age was 69 years (range 50–80). Performance status was 0 (n = 7), 1 (n = 11) and 2 (n = 7) and the majority, 17 patients, had metastatic disease compared to eight with locally advanced, unresectable disease.
Chemotherapy was given for a median of five full months (range 0–14) and dose modification for toxicity, mainly neurotoxicity, occurred in 80% of the patients, while postponement was necessary in five cases. One grade 4 (febrile neutropenia) and six grade 3 toxicity (febrile neutropenia, hyperglycemia and urticaria) were observed, while the most frequent grade 1–2 toxicities were neurotoxicity (17), nausea or vomiting (16), pain (14), constipation (11) and diarrhea (7).
Efficacy
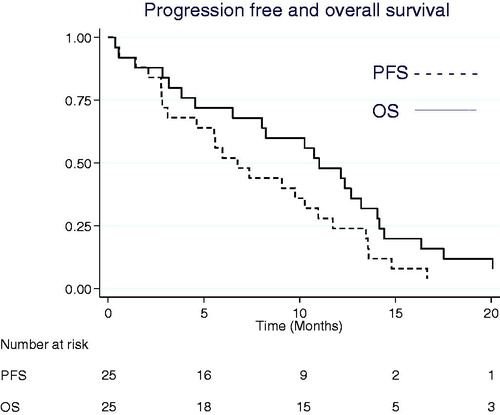
Among 17 patients evaluable for response, one complete and two partial responses were seen and the rest had stable disease as best response. Median PFS was 6.8 months (95% CI 3.1–11.0). Two patients are still alive after more than three and six years, respectively. Median overall survival was 11.2 months (95% CI 6.6–14.3) (). Thirteen patients were alive without progression at the evaluation after six months. Thus, the primary endpoint, fraction of PFS at six months, was 52% (95% CI 31–69%).
Figure 1. Kaplan-Meier estimates for overall survival (OS) and progression-free survival (PFS).
Plasma DNA
Plasma samples were available from 24 of 25 patients. The median of total plasma DNA was 6912 (range 915–131896) gene copies per mL. In 13 patients, the known tumor tissue KRAS mutation was detectable at baseline. Patients with a higher level of total plasma DNA and patients with detectable KRAS mutation in plasma tended to have inferior PFS and OS as shown in , with a statistical significant shorter OS for patients with total plasma DNA above the median.
Table 1. Progression free survival (PFS) and overall survival (OS) is shown for 24 patients with a level of total plasma DNA below or above the median and also for patients with (n = 13) or without (n = 11) detectable KRAS mutation.
KRAS mutation rate
In the study period, 213 patients had a successful KRAS mutation test and initiated systemic therapy. Most of the patients were included in clinical trials [Citation6] and the rest did not fulfill eligibility criteria. A total of 38 patients (17.8%) had tumors with KRAS mutations with a median survival of 9.8 months (95% CI 8.7–11.5) compared to 9.7 months (95% CI 6.6–12.6) for wild-type (p = .35).
Discussion
This phase 2 study is the first to test triple chemotherapy in a uniform population of patients with KRAS mutant BTC. The data supports the feasibility of patient selection even in a rare disease and rare mutations. The study met its primary endpoint with 13 patients (52%) alive and without progression at six months.
KRAS exon 2 mutations were expected in half of the patients [Citation13,Citation14], but were only found in 18% in accordance with later reports [Citation15]. We found no clinically relevant differences in survival of patients undergoing chemotherapy based on the presence KRAS mutation, but the trial design did not allow separation of the predictive and prognostic entities of this biomarker.
Phase III data about combination chemotherapy in advanced BTC is limited to one large and well conducted trial where gemcitabine combined with cisplatin was superior compared to gemcitabine monotherapy [Citation16]. Triple chemotherapy has numerically a comparable efficacy in phase II trials [Citation17–19]. Median OS in the present trial was 11.2 months and thus comparable to both published phase II and III trials even though the rate of patients with poor performance score was higher, 28% vs. 12% in the phase III trial. Interestingly, two patients with metastasis are still alive. One without progression after 7 years and the other had reintroduction of the chemotherapy after 3 years and is now progression free after further four years.
The analysis of plasma DNA, liquid biopsies, may be a future biomarker. In exploratory analyses we found a significantly improved survival in patients with at low level of total plasma DNA. The potential clinical use is further supported by trends toward improved PFS and by trends toward improved outcomes for patients without detectable KRAS mutation in plasma.
Strengths of the study are the trial design for precision medicine in a rare disease and screening data allowing elaboration of the potential clinical value of KRAS codon 2 mutations. Weaknesses are the limited number of patients with this subtype of BTC, the non-randomized design and mutation analysis limited to KRAS codon 2.
Future research should explore broad genomic profiling and molecular subclassification [Citation20]. Furthermore, a high level of plasma DNA may be evaluated as a marker for identifying patients with short survival where supportive care could be suggested instead of chemotherapy.
Conclusions
Pretreatment molecular characterization was feasible in BTC, but the rate of KRAS mutations was low. The study met its primary endpoint with a fraction of PFS at six months of 52%. The effect of combination chemotherapy with gemcitabine, oxaliplatin and capecitabine in this selected population was comparable to results from unselected groups with PFS and OS of 6.8 and 11.2 months, respectively.
Supplemental Material
Download PDF (103.3 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Jepsen P, Vilstrup H, Tarone RE, et al. Incidence rates of intra- and extrahepatic cholangiocarcinomas in Denmark from 1978 through 2002. J Natl Cancer Inst. 2007;99(11):895–897.
- Hezel AF, Zhu AX. Systemic therapy for biliary tract cancers. Oncologist. 2008;13(4):415–423.
- Lievre A, Bachet J-B, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995.
- Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. JCO. 2008;26(10):1626–1634.
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310.
- Jensen LH, Lindebjerg J, Ploen J, et al. Phase II marker-driven trial of panitumumab and chemotherapy in KRAS wild-type biliary tract cancer. Ann Oncol. 2012;23(9):2341–2346.
- Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–216.
- Trotti A, Colevas A, Setser A, et al. CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol. 2003;13(3):176–181.
- Steffensen KD, Waldstrøm M, Grove A, et al. Improved classification of epithelial ovarian cancer: results of 3 Danish cohorts. Int J Gynecol Cancer. 2011;21(9):1592–1600.
- Pallisgaard N, Spindler K-L, Andersen RF, et al. Controls to validate plasma samples for cell free DNA quantification. Clin Chim Acta. 2015;446:141–146.
- Thomsen CB, Hansen TF, Andersen RF, et al. Monitoring the effect of first line treatment in RAS/RAF mutated metastatic colorectal cancer by serial analysis of tumor specific DNA in plasma. J Exp Clin Cancer Res. 2018;37(1):55.
- Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials. 1989;10(1):1–10.
- Tannapfel A, Benicke M, Katalinic A, et al. Frequency of p16(INK4A) alterations and K-ras mutations in intrahepatic cholangiocarcinoma of the liver. Gut. 2000;47(5):721–727.
- Tannapfel A, Sommerer F, Benicke M, et al. Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut. 2003;52(5):706–712.
- Chang Y-T, Chang M-C, Huang K-W, et al. Clinicopathological and prognostic significances of EGFR, KRAS and BRAF mutations in biliary tract carcinomas in Taiwan. J Gastroenterol Hepatol. 2014;29(5):1119–1125.
- Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362(14):1273–1281.
- Yamashita Y, Taketomi A, Itoh S, et al. Phase II trial of gemcitabine combined with 5-fluorouracil and cisplatin (GFP) chemotherapy in patients with advanced biliary tree cancers. Jpn J Clin Oncol. 2010;40(1):24–28.
- Wagner AD, Buechner-Steudel P, Moehler M, et al. Gemcitabine, oxaliplatin and 5-FU in advanced bile duct and gallbladder carcinoma: two parallel, multicentre phase-II trials. Br J Cancer. 2009;101(11):1846–1852.
- Lassen U, Jensen LH, Sorensen M, et al. A phase I-II dose escalation study of fixed-dose rate gemcitabine, oxaliplatin and capecitabine every two weeks in advanced cholangiocarcinomas. Acta Oncol. 2011;50(3):448–454.
- Jusakul A, Cutcutache I, Yong CH, et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 2017;7(10):1116–1135.