
Abstract
Background: We have reported that BRAF V600E mutations and microsatellite instability-high (MSI-H) are more prevalent in a population-based cohort of metastatic colorectal cancer (mCRC) patients than has been reported from clinical trials or hospital-based patient groups. The aim was to explore if other mutations in mCRC differ in prevalence between these cohorts in relation to mismatch repair status and primary tumor location and if presence of bone or brain metastases is associated with any mutations.
Material and methods: A population-based cohort of 798 mCRC patients from three regions in Scandinavia was used. Forty-four cancer related genes were investigated in a custom designed Ampliseq hotspot panel. Differences in survival were analyzed using the Kaplan–Meier estimator and the Cox regression analysis.
Results: Determination of mutations was possible in 449/501 patients for 40/44 genes. Besides BRAF V600E, seen in 19% of the tumors, none of the other mutations appeared more prevalent than in trial cohorts. BRAF V600E and MSI-H, seen in 8%, were associated with poor prognosis as was right-sided primary tumor location (39%) when compared to left-sided and rectum together; however, in a multivariable regression, only the BRAF mutation retained its statistical significance. No other mutations were associated with poor prognosis. ERBB2 alterations were more common if bone metastases were present at diagnosis (17% vs. 4%, p = .011). No association was found for brain metastases. Fifty-two percent had an alteration that is treatable with an FDA-approved targeted therapy, chiefly by EGFR-inhibitor for RAS wild-type and a check-point inhibitor for MSI-H tumors.
Conclusions: Right-sided tumor location, BRAF V600E mutations, but no other investigated mutation, and MSI-H are more commonly seen in an unselected cohort than is reported from clinical patient cohorts, likely because they indicate poor prognosis. Half of the patients have a tumor that is treatable with an already FDA-approved targeted drug for mCRC.
Introduction
Colorectal cancer (CRC) is the third most prevalent cancer globally [Citation1]. Previous studies have sought to discover prognostic and predictive somatic mutations for novel treatments in CRC by genomic sequencing [Citation2,Citation3]. This is particularly important for metastatic CRC (mCRC) patients that have the poorest survival, median between 10 and 12 months in the general population and up to 30 months in selected patient groups [Citation4,Citation5]. It is important that exploratory studies not only focus on the fittest patients, i.e., those suitable for trial inclusion, but instead look at the entire disease population.
We have shown that BRAF V600E mutations [Citation6] and microsatellite instability-high (MSI-H) [Citation7] are more common in an unselected population of Scandinavian patients with mCRC than in patient groups derived from clinical trials or specialized hospitals (21% BRAF [Citation6] mutated in the population vs. 5–12% [Citation8–10]; and 7% MSI-H [Citation7] vs. 3–4% [Citation10,Citation11]). A likely reason for this is that patients with tumors harboring BRAF mutations or MSI-H have a poor prognosis with short survival [Citation12,Citation13]; they often fail to be included in trials or are not referred to specialized hospitals. For decades, the generally held view was that the primary tumor location was not important beyond separating colon from rectum. However, this has recently emerged as a prognostic factor in mCRC and as a predictive factor for treatment with epidermal growth factor receptor (EGFR) inhibitors [Citation14]. In a meta-analysis of 14 first-line studies, the proportion of right-sided tumors varied between 18 and 36% [Citation15]. Several reports claim that right-sided tumors have a worse prognosis and require different treatment upfront, beyond the information provided by investigation of RAS, BRAF and microsatellite instability (MSI). However, most of this evidence has been obtained from trial patients and real-world evidence is limited [Citation16].
The primary purpose of this study was to explore whether mutation prevalence, in known cancer genes in an unselected population differs from that reported in trial populations, and if the location of the primary tumor is prognostic in a population-based cohort. Further, the unselected material allows exploration of mutations related to two uncommon metastatic sites in mCRC, bone and brain, which are frequently underrepresented in clinical trials. Finally, we wanted to examine how frequent other molecular changes of potential interest for targeted therapy are in a population-based material.
Materials and methods
Patient cohort
The cohort represents an unselected population of all non-resectable mCRC patients diagnosed in three regions in Scandinavia (Uppsala, Sweden; Odense, Denmark; Bergen, Norway), with an mCRC diagnosis between October 2003 and August 2006 [Citation17]. An informed consent was signed by all patients expect for 49 patients identified from regional cancer registries [Citation6]. All information was prospectively collected from the clinical records by clinicians and research nurses, subsequently anonymized and de-identified before analysis. Regional ethical committees in Norway, Sweden and Denmark approved the study as well permission to include patients not prospectively identified to make the cohort truly population-based.
Tissue retrieval, tissue microarray generation and DNA extraction
Hematoxylin–eosin stained slides from primary tumors or metastases were examined so representative tumor parts could be selected from the corresponding tissue blocks, and non-necrotic tumor areas with few other cells admixed marked. Tissue microarray (TMA) generation and DNA extraction were performed using 1 mm tissue cores from the original primary tumor block except in six patients that were from metastatic lesions. The Beecher Instruments Manual Tissue Arrayer MTA-1 was used to generate TMAs. DNA was recovered from 505 (63%) tissue cores by Recoverall Total Nucleic Acid Isolation kit (Ambion, Austin, TX, USA). The remaining cases had either no remaining cancer tissue or not enough material to take cores for research; these were all diagnosed using small colorectal biopsies or needle biopsies of metastatic lesions.
Microsatellite instability analysis
MSI status was determined via a combination of immunohistochemistry (IHC) and polymerase chain reaction (PCR) techniques, see Supplementary Methods.
ERBB2/HER2 IHC and dual-color silver-enhanced in situ hybridization (SISH)
Tumors were stained with a monoclonal antibody against human HER2, clone CL0268 (mouse), dilution 1:250 (Atlas Antibodies, Stockholm, Sweden). Protein expression was ranked from 1+ (weak intensity) to 3+ (strong intensity). Bright-field dual-color SISH analysis was performed for all TMAs using an automatic SISH staining device BenchMark ULTRA, according to manufacturer’s instructions for INFORM HER2 DNA and INFORM Chromosome 17 (CEP17) probes (Ventana Medical Systems, Tucson, AZ, USA). HER2/CEP17 SISH signals were counted for all 2+ and 3+ IHC scored samples according to the guidelines for staining of gastric cancers. All samples with a HER2/CEP17 ratio ≥2.0 were considered to have amplified ERBB2 expression. Adjacent benign cells were used as controls. HER2 status was also determined accordingly to HERACLES diagnostic criteria for CRC [Citation18].
Targeted sequencing and data analysis
A custom designed Ampliseq hotspot panel (Thermo-Fisher Scientific, Waltham, MA, USA) covering 194 amplicons from 44 cancer related genes was designed using Ion AmpliSeq Designer (Supplementary Table 1). The AmpliSeq panel was designed using specific settings customized for FFPE samples. Sequencing libraries were prepared from 10 ng of genomic DNA according to the Ion AmpliSeq Library Kit 2.0 user guide and quantified using the Agilent Bioanalyzer instrument and the Agilent High Sensitivity DNA kit. Emulsion PCR, enrichment and chip loading were performed on the Ion Chef system using the Ion PI Chef Kit (Thermo Fisher Scientific, Waltham, MA, USA). The samples were sequenced on the Ion Proton System using an Ion PI chip and 200 bp chemistry. Data analysis and variant calling were performed as described in Supplementary Methods. Somatic alterations were defined in tiers according to OncoKB levels of evidence [Citation19] and presented in a figure made with Oncoprinter [Citation20,Citation21].
Statistical analyses
Fisher’s exact test was performed for group comparisons and p value < .05 was considered statistically significant. Overall survival (OS) was the time between the dates of diagnosis of metastatic disease and death or censored for patients alive in February 2014. The Kaplan–Meier estimator and the Cox multiple regression were used for OS analysis. Statistical analyses were performed using R software, version 3.6.0 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Cohort and mutation characterization
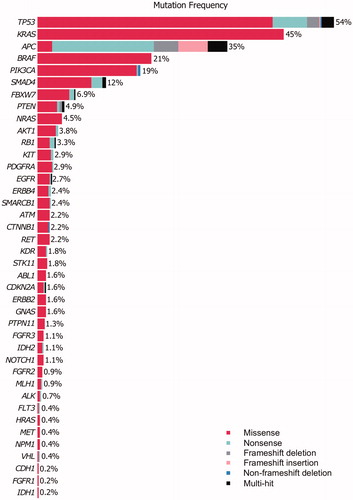
The study included 798 patients, of which 701 patients had surgical specimens with invasive cancer. Tissue cores could be generated from 505 patients (Supplementary Figure 1). The sequenced patients were representative of the entire cohort, by age and sex distribution, and primary tumor location (Supplementary Table 2). Sequencing data were obtained from 501 tumors at a median amplicon coverage per tumor of 4,801. EZH2 had the highest amplicon coverage (median 19,084) while PTEN had the lowest coverage (median 2,194) (Supplementary Table 3). Due to the high overall coverage, we used a stringent cutoff of 1,000-fold average coverage; 95% (476/501) of the samples met this criterion. After applying the filters described in Supplementary Methods, 411/449 (92%) patients carried at least one mutation in 40/44 genes sequenced (EZH2, HNF1A, MPL and SRC had no mutations). In total, 1,249 somatic nonsynonymous single-nucleotide variants and indels were identified (40 frameshift deletions, 33 frameshift insertions, four non-frameshift deletions, 984 missense and 188 nonsense point mutations), corresponding to 437 unique and 142 recurrent mutations within the set. The most recurrent hotspot mutation was BRAF V600E (86 cases) followed by KRAS G12D and G12V with 60 and 40 cases, respectively. From the gene panel, 6/44 genes were mutated in more than 10% of the patients, namely TP53 (242/449; 54%), KRAS (201/449; 45%), APC (155/449; 35%), BRAF (93/449; 21%), PIK3CA (84/449; 19%) and SMAD4 (56/449; 12%) (). To identify significantly mutated genes, we fit a linear regression model using the total number of sequenced base pairs and total number of mutations for each gene. Ten genes, KRAS, BRAF, TP53, APC, CTNNB1, PIK3CA, AKT1, NRAS, SMAD4 and FBXW7 had a higher mutation rate than expected by chance (Supplementary Figure 2).
Figure 1. Frequency of altered genes in a Scandinavian unselected cohort of metastatic colorectal cancer by type of mutation.
Microsatellite instability
By combining the data from IHC and PCR genotyping, we divided the cohort in MSI-H (36/449; 8%) and MSS/MSI-L phenotypes (413/449; 92%), referred to as MSS. As expected, MSI-H patients were predominately females with poorly or undifferentiated tumors located in the right colon () [Citation11]. They differed in metastatic pattern from MSS tumors with fewer liver (36% vs. 66%, p < .001) and lung (6% vs. 26%, p = .004) metastases but more lymph node metastases (53% vs. 28%, p = .004). Overall, 64% of the patients received chemotherapy with no significant difference between MSI-H and MSS tumors. OS was shorter in patients with MSI-H tumors (6 vs. 13 months, p = .006), even when only considering patients receiving chemotherapy (11 vs. 19 months, p = .013, ). Complete or partial response to first-line treatment was higher for the MSS cases (43% vs. 5%, p < .001, ). As expected, the MSI-H patients had a higher BRAF mutation prevalence (75% vs. 16%, p < .001, Supplementary Table 4) and OS was shorter if the tumor was BRAF mutated (). From the significantly mutated genes, only APC (37% vs. 11%, p = .002) and KRAS (48% vs. 11%, p < .001) had significantly higher mutation prevalence in MSS compared to MSI-H tumors (Supplementary Table 4). Aside from BRAF, no other significantly mutated gene was associated with poor prognosis even when considering first-line treated patients only (Supplementary Figure 3).
Figure 2. The Kaplan–Meier analysis of overall survival (OS) for all patients and for patients given 1st-line chemotherapy treatment according to MSI status, BRAF mutation and primary tumor location. p value was calculated with log-rank test. (A) OS for all patients and (B) for patients given 1st-line chemotherapy by MSI status. (C) OS for all patients and (D) for patients given 1st-line chemotherapy by BRAF mutation status. (E) OS for all patients and (F) for patients given 1st-line chemotherapy by primary tumor location.
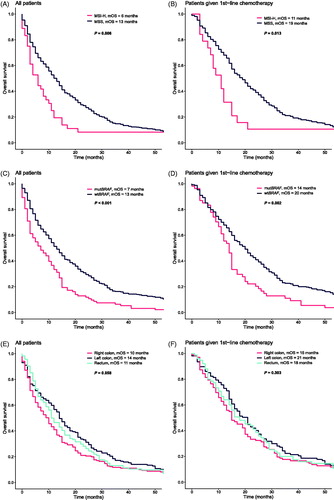
Table 1. Comparison of patient and tumor characteristics between patients with MSS and MSI-H tumors.
Gene alterations according to primary tumor location, age and selected metastatic sites
In the cohort of 449 patients with sequenced tumors, 38% were right-sided (right colon and transversum), 35% left-sided (left colon and sigmoideum) and 25% were rectal tumors. The patients with right-sided tumors were more likely to be older, female and have poorly differentiated tumors with lymph node and peritoneal metastases (). The MSI-H phenotype was more frequent in right-sided tumors (18% vs. 1% and 3% for left-sided and rectal tumors, respectively, p < .001). Similarly, BRAF mutation frequency was higher in right-sided tumors but decreased throughout the left colon and rectum (38% vs. 14% vs. 5%, p < .001). Also, the PIK3CA mutation frequency was highest in the right colon, followed by rectal tumors and lowest in the left colon (24% vs. 17% vs. 13%, p = .044). No other gene mutation prevalence differed significantly by primary tumor location. No gene mutation prevalence, aside from AKT1, was influenced by age (Supplementary Table 5). In all patients (and in patients treated with chemotherapy, for choice of chemotherapy, see Supplementary Figure 4), no significant differences could be detected for OS according to tumor sidedness based on MSI expression and BRAF mutation status (; ). When right-sided colon tumors were compared with left-sided colon and rectum tumors together, median OS was 10 months vs. 14 months for all patients (p = .046) and 15 months vs. 18.5 months (p = .270) for patients that received first-line treatment. In a multiple Cox regression analysis, including MSI status, BRAF mutation status, primary tumor location (right colon vs. left colon and rectum together) and whether the patient received first-line treatment, only BRAF mutation was significantly associated with reduced OS, while receiving first-line treatment was associated with an increased OS (Supplementary Figure 5).
Table 2. Comparison of patient characteristics and altered genes by primary tumor location (seven patients had multiple sites).
Patients with bone metastases at time of diagnosis of metastatic disease (n = 30, 7%) more often had a rectal primary (54%) than a colon primary (29% right-sided and 18% left-sided, p = .004), and more often lung (p = .049) and multiple site metastases (p < .001), but less often liver metastases (p = .009). They also received radiotherapy more often than patients without simultaneous bone metastases (57% vs. 7%, p < .001). Patients with bone metastases had poorer performance status and their OS was significantly shorter (8 vs. 12 months, p = .015). None of the patients with simultaneous bone metastases had MSI-H tumors, but two (6%) patients with MSI-H tumors later developed bone metastases. Thirty-nine (10%) patients with MSS tumors without simultaneous bone metastases later developed bone metastases. Furthermore, ERBB2 alteration was more often seen in patients with simultaneous bone metastasis (17% vs. 4%, p = .011, Supplementary Table 6).
No patient had brain metastases at the time of diagnosis of their primary tumor. Of the 26 patients who developed brain metastases after a median of 20 months (range 1–139), one (3%) developed in a patient with an MSI-H tumor and 25 (6%, p = .711) in patients with MSS tumors. Lung metastases were more common for the patients with brain metastases (54% vs. 23%, p = .001) and no gene alteration frequency differed between these groups (Supplementary Table 7).
Clinically actionable alterations
KRAS was the most altered oncogene with 45% of the tumors having at least one oncogenic alteration. Five MSS patients had two co-occurring KRAS mutations, where one of them had two known oncogenic mutations (G13D and A18T with variant allele frequency (VAF) of 12%, Supplementary Figure 6A). NRAS was altered in 4.5% of the cases. BRAF was mutated in 21% of the cohort and the most common BRAF alteration was V600E, observed in 19% of tumors. One MSS patient had a complex substitution (c.1798_1799GT > AG) in BRAF that led to a rare V600R alteration. Non-V600 BRAF mutations were present in six cases and further classified according to [Citation22]; G469R, a class 2 mutation leading to intermediate kinase activity; G466E (two cases), D594G and D594N, class 3 alterations leading to no kinase activity; and V590I of undefined non-V600 class described in one previous salivary gland tumor [Citation23]. Two of the class 3 alterations, observed in colon cancers were co-mutated with KRAS, while two were present in rectal cancers without KRAS co-mutation. Two patients had two co-occurring BRAF mutations, where one of them presented two neighboring oncogenic mutations (V600E and K601N with VAF of 33% and 26%, respectively, Supplementary Figure 6B). PIK3CA was altered in 84 (19%) patients. In total, 15 (3.3%) patients were scored 2+ or 3+ by IHC and had a positive SISH analysis for ERBB2 according to the breast and gastric cancer criteria, while 12 (2.6%) had this when considering HERACLES CRC diagnostic criteria. One-third of the patients had a co-occurring KRAS mutation irrespective of what criteria was used (Supplementary Table 8). ERBB2 mutation was found in seven additional cases.
Discussion
Numerous studies have explored the genomic and phenotypical heterogeneity in mCRC, but almost exclusively from selected trial or hospital-based patient series not representative of the general population. We evaluated genomic properties in almost 450 mCRC tumors from an unselected population-based cohort where all diagnosed individuals with mCRC not immediately possible to resect were identified. We could substantiate that BRAF V600E mutations and MSI-H are about twice as common as in previously reported datasets. While other studies commonly report about 6–8% (range 5–12%) BRAF V600E mutations [Citation24] and 3–4% MSI-H tumors [Citation11], this study revealed 19% BRAF V600E mutations and 8% MSI-H. The likely explanation for this is the poor prognosis of these patient groups; therefore, they are underrepresented in patient materials from clinical trials or hospital-based cohorts where patients must fulfill certain inclusion. The lower than expected mutation frequency in two genes, APC and TP53, 35% vs. expected 80% and 54% vs. expected 65%, respectively can likely be explained by incomplete coverage of these genes in the gene panel design where only the main mutation driver cluster regions were covered as previously published by Overman et al. [Citation25].
None of the other known somatic mutations of potential interest in mCRC, including PIK3CA mutations and ERBB2 expression, were more prevalent in this patient group compared to other cohorts, indicating that the prognosis for those groups is not particularly poor. Since both BRAF V600E mutation and MSI-H phenotype are indicators of poor prognosis and poor response to conventional chemotherapy, there is an urgent need to determine these properties at the time of diagnosis. The very poor prognosis for patients with MSI-H tumors is likely a combination of a more aggressive tumor and a poorer response to therapy. The very poor response to first-line treatment in patients with MSI-H tumors reported here, underlines the importance of evaluating immunotherapy upfront [Citation26].
In contrast to most recent studies in mCRC, this study could not substantiate worse survival for right-sided compared to left-sided tumors [Citation27,Citation28]. In this study and a Canadian population study [Citation16], we find that about 40% of patients with mCRC have a right-sided tumor. The two mCRC subgroups with the worst prognosis, MSI-H and BRAF-mutated tumors are all more common in right-sided tumors. Whether the poor prognosis of right-sided tumors is solely caused by these molecular properties is still an unresolved question, but it has been suggested that even if there is no overactivation of the MAPK signaling pathway, right-sided tumors should not be treated with EGFR-inhibitors [Citation14]. The Canadian study also noticed poorer prognosis for treated patients (47% received chemotherapy) with right-sided tumors but did not report any molecular analyses. Interestingly, in this cohort, BRAF and PIK3CA were the only genes with significantly higher mutation prevalence in right-sided tumors, as previously reported [Citation27,Citation29]. However, we did not observe that any other gene was associated with sidedness, which could be due to the unselected nature of this cohort.
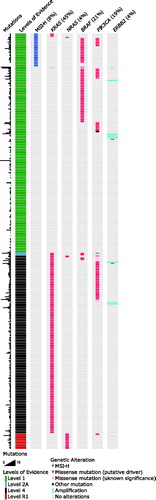
Wild-type KRAS and NRAS are FDA-approved biomarkers for administration of anti-EGFR antibodies [Citation30–32], and 51% (230/449) of the cases in this study were wild-type for both genes. Further, BRAF and PIK3CA mutations or ERBB2 amplifications can also result in resistance to EGFR-inhibition [Citation33] meaning that only about half (126/230) of the RAS wild-type patients are ideal candidates for this treatment. ERK and MEK inhibitors are studied as targeted drugs for RAS oncogenic mutations [Citation34], as well as AMG 510 (NCT03600883), a novel small molecule KRAS G12C inhibitor potentially suitable for 2% (8/449) of our cohort. In mCRC, there is compelling clinical evidence for the use of encorafenib and cetuximab with binimetinib in patients with BRAF V600E mutation, meaning this could be a possible therapy for 19% (87/449) of this cohort [Citation35]. In vitro evidence supports the use of PLX8394 for BRAF non-V600 alterations [Citation36], present in five (1%) patients in this study. ERBB2 amplification assessment is routinely done in breast and esophagogastric cancers as these tumors can be targeted by FDA-approved drugs. We could detect ERBB2 amplification with SISH analysis in 3% (15/449) of the patients, translating to the percentage of mCRC patients that could benefit from targeted therapy. Increasing evidence has led to FDA approval of the use of immune checkpoint inhibitors in patients with MSI-H tumors, representing 8% (36/449) of our population [Citation37]. To summarize, 53% (237/449) of our cohort could have potential benefit from an FDA-approved targeted therapy in mCRC, mainly based on the absence of overactive RAS-MAPK signaling ().
Figure 3. Distribution of patients with an FDA-approved (237/449, 53%) or potentially actionable alteration in an unselected cohort of metastatic colorectal cancers. Total amount of mutations, OncoKB levels of evidence, MSI status and mutation status of selected genes are represented in different columns for all patients. Each row represents one patient. Color coding indicates the type of event in each column. Patients in rows indicated with level 1 evidence have at least one FDA-approved biomarker that responds to an FDA-approved drug (RAS wild-type, EGFR-inhibitor; MSI-H, checkpoint inhibitor); patients in level 2A have at least one standard of care biomarker predicted to respond to an FDA-approved drug (BRAF V600E, BRAF-inhibitor); those in level 4 have a biomarker with predictive compelling biological evidence to respond to a drug (KRAS mutation, MEK-inhibitor) and level those in R1 have a standard care biomarker that is resistant to an FDA-approved drug (NRAS mutation, EGFR-inhibitor).
In conclusion, aside from MSI-H phenotype and BRAF V600E mutations, no other molecular change was more, or less, prevalent than in selected cohorts. Both MSI-H and BRAF mutation are associated with poor prognosis, potentially explaining why these features are more prevalent than has previously been reported [Citation8–11]. This may also explain why there is no difference in mutation prevalence for other genes included in this study, as no other mutation was associated with poor prognosis. However, the limited numbers for most of those mutations prevent us from making firm conclusions. Besides alterations in ERBB2, seen more frequently if bone metastases were present, we could not identify alterations associated with the rarer metastatic sites of bone or brain. Similar to previous studies, a rectal primary was more often associated with bone metastatic sites than colon cancer [Citation38]. Finally, and opposed to a generally held view [Citation27,Citation28], we could not substantiate that right-sided tumors that do not harbor a BRAF mutation or an MSI-H phenotype have a worse outcome than left-sided tumors, whether receiving chemotherapy or not.
The patient material collected from 2003 to 2006 and the comparatively short OS could make one question the relevance of using this study to draw conclusions that are applicable to today’s mCRC diagnosis and prognosis. However, the tumor panorama has not changed, and the OS reflects the population-based nature of the material. As such, it is likely unique. The median OS of 9 months (7.8–10.2) is heavily influenced by the non-actively treated patients (43% of all patients), due to their old age, presence of severe co-morbidities or very aggressive disease with poor performance status. The comparatively low median OS of 15 months (13.4–16.6) in the chemotherapy group can be explained by (i) the 22% that only received single fluoropyrimidine, (ii) the inclusion of patients with co-morbidities and laboratory abnormalities that disqualify them from trial participation and (iii) the exclusion of patients with upfront resectable metastatic disease. For patients eligible for trial participation, all three active cytotoxic drugs, bevacizumab and the EGFR-inhibitor cetuximab were available and used, resulting in a median OS of about 24 months [Citation17], in line with what has been reported from clinical trials including mCRC patients [Citation30,Citation31,Citation39]. The up to 30 months median OS reported in recent trials [Citation4] reflects molecular selection and the inclusion of patients having resectable metastatic disease. Thus, we believe our prognostic associations are relevant for today’s patients. Taken together, we present unbiased mutation frequencies in mCRC and estimate the true percentage of patients potentially eligible for targeted treatment in an unselected western-world mCRC cohort.
| Abbreviations | ||
| CRC | = | colorectal cancer |
| mCRC | = | metastatic colorectal cancer |
| MSI-H | = | microsatellite instability-high |
| EGFR | = | epidermal growth factor receptor |
| MSI | = | microsatellite instability |
| TMA | = | tissue microarray |
| IHC | = | immunohistochemistry |
| PCR | = | polymerase chain reaction |
| MSS | = | microsatellite stability |
| MSI-L | = | microsatellite instability-low |
| SISH | = | silver-enhanced in situ hybridization |
| CEP17 | = | INFORM Chromosome 17 |
| OS | = | overall survival |
| VAF | = | variant allele frequency |
Supplemental Material
Download Zip (809.9 KB)Disclosure statement
The authors declare no conflicts of interest.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017: Colorectal Cancer Statistics, 2017. CA Cancer J Clin. 2017;67(3):177–193.
- Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–274.
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337.
- Van Cutsem E, Cervantes A, Adam R, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27(8):1386–1422.
- Glimelius B, Cavalli-Björkman N. Metastatic colorectal cancer: current treatment and future options for improved survival. Medical approach – present status. Scand J Gastroenterol. 2012;47(3):296–314.
- Sorbye H, Dragomir A, Sundström M, et al. High BRAF mutation frequency and marked survival differences in subgroups according to KRAS/BRAF mutation status and tumor tissue availability in a prospective population-based metastatic colorectal cancer cohort. PLoS One. 2015;10(6):e0131046.
- Aasebø KØ, Dragomir A, Sundström M, et al. Consequences of a high incidence of microsatellite instability and BRAF‐ mutated tumors: a population‐based cohort of metastatic colorectal cancer patients. Cancer Med. 2019;8(7):3623–3635.
- Tveit KM, Guren T, Glimelius B, et al. Phase III trial of cetuximab with continuous or intermittent fluorouracil, leucovorin, and oxaliplatin (Nordic FLOX) versus FLOX alone in first-line treatment of metastatic colorectal cancer: the NORDIC-VII Study. J Clin Oncol. 2012;30(15):1755–1762.
- Yaeger R, Cercek A, Chou JF, et al. BRAF mutation predicts for poor outcomes after metastasectomy in patients with metastatic colorectal cancer: metastasectomy in BRAF-Mutant mCRC. Cancer. 2014;120(15):2316–2324.
- Venderbosch S, Nagtegaal ID, Maughan TS, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res. 2014;20(20):5322–5330.
- Koopman M, Kortman GAM, Mekenkamp L, et al. Deficient mismatch repair system in patients with sporadic advanced colorectal cancer. Br J Cancer. 2009;100(2):266–273.
- Tran B, Kopetz S, Tie J, et al. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer: metastatic pattern in BRAF mutant CRC. Cancer. 2011;117(20):4623–4632.
- Fujiyoshi K, Yamamoto G, Takenoya T, et al. Metastatic pattern of stage IV colorectal cancer with high-frequency microsatellite instability as a prognostic factor. Anticancer Res. 2017;37(1):239–248.
- Arnold D, Lueza B, Douillard J-Y, et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann Oncol. 2017;28(8):1713–1729.
- Holch JW, Ricard I, Stintzing S, et al. The relevance of primary tumour location in patients with metastatic colorectal cancer: a meta-analysis of first-line clinical trials. Eur J Cancer. 2017;70:87–98.
- Ahmed S, Pahwa P, Le D, et al. Primary tumor location and survival in the general population with metastatic colorectal cancer. Clin Colorectal Cancer. 2018;17(2):e201–e206.
- Sorbye H, Pfeiffer P, Cavalli-Björkman N, et al. Clinical trial enrollment, patient characteristics, and survival differences in prospectively registered metastatic colorectal cancer patients. Cancer. 2009;115(20):4679–4687.
- Valtorta E, Martino C, Sartore-Bianchi A, et al. Assessment of a HER2 scoring system for colorectal cancer: results from a validation study. Mod Pathol. 2015;28(11):1481–1491.
- Chakravarty D, Gao J, Phillips S, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017;1:1–16.
- Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1.
- Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data: figure 1. Cancer Discov. 2012;2(5):401–404.
- Yao Z, Yaeger R, Rodrik-Outmezguine VS, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature. 2017;548(7666):234–238.
- Tetsu O, Phuchareon J, Chou A, et al. Mutations in the c-Kit gene disrupt mitogen-activated protein kinase signaling during tumor development in adenoid cystic carcinoma of the salivary glands. Neoplasia. 2010;12(9):708–717.
- Misale S, Di Nicolantonio F, Sartore-Bianchi A, et al. Resistance to anti-EGFR therapy in colorectal cancer: from heterogeneity to convergent evolution. Cancer Discov. 2014;4(11):1269–1280.
- Overman MJ, Morris V, Kee B, et al. Utility of a molecular prescreening program in advanced colorectal cancer for enrollment on biomarker-selected clinical trials. Ann Oncol. 2016;27(6):1068–1074.
- Oliveira AF, Bretes L, Furtado I. Review of PD-1/PD-L1 inhibitors in metastatic dMMR/MSI-H colorectal cancer. Front Oncol. 2019;9:396.
- Yaeger R, Chatila WK, Lipsyc MD, et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell. 2018;33(1):125–136.e3.
- Loupakis F, Yang D, Yau L, et al. Primary tumor location as a prognostic factor in metastatic colorectal cancer. J Natl Cancer Inst. 2015;107:pii: dju427.
- Loree JM, Pereira AAL, Lam M, et al. Classifying colorectal cancer by tumor location rather than sidedness highlights a continuum in mutation profiles and consensus molecular subtypes. Clin Cancer Res. 2018;24(5):1062–1072.
- Van Cutsem E, Köhne C-H, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360(14):1408–1417.
- Douillard J-Y, Siena S, Cassidy J, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28(31):4697–4705.
- Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303–312.
- Therkildsen C, Bergmann TK, Henrichsen-Schnack T, et al. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: a systematic review and meta-analysis. Acta Oncol. 2014;53(7):852–864.
- Sullivan RJ, Infante JR, Janku F, et al. First-in-class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018;8(2):184–195.
- Kopetz S, Grothey A, Yaeger R, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med. 2019;381(17):1632–1643.
- Tutuka CSA, Andrews MC, Mariadason JM, et al. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol Cancer. 2017;16(1):112.
- Overman MJ, Lonardi S, Wong KYM, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36(8):773–779.
- Riihimäki M, Hemminki A, Sundquist J, et al. Patterns of metastasis in colon and rectal cancer. Sci Rep. 2016;6:29765.
- Renouf DJ, Lim HJ, Speers C, et al. Survival for metastatic colorectal cancer in the bevacizumab era: a population-based analysis. Clin Colorectal Cancer. 2011;10(2):97–101.