
Introduction
T-prolymphocytic leukemia (T-PLL) is a rare, mature T-cell leukemia representing less than 2% of mature lymphocytic leukemias [Citation1,Citation2]. T-PLL is generally resistant to conventional chemotherapy with a median survival of approximately 7 months. T-PLL primarily affects elderly adults with a median age at onset of 63 years, however in patients with ataxia telangiectasia T-PLL accounts for 3% of all malignancies with a much earlier onset (median age 30 years) [Citation3]. We report a case of a 26-year-old male with alemtuzumab-refractory T-PLL 16 years after curatively treated T-cell acute lymphoblastic leukemia (T-ALL). Partly successful combination treatment with venetoclax, alemtuzumab, vorinostat, and cladribine is discussed along with clinical, morphological, immunophenotypic, molecular, genetic, and cytogenetic features.
Case report
A 26-year-old man presented with a few months of progressive weakness and fatigue. Main complaints were a few days of dizziness and tiredness as well as weeks of diarrhea. The patient had a history of T-ALL 16 years prior, where complete remission was achieved following combination therapy of anthracycline, cyclophosphamide, methotrexate, asparaginase, cytarabine, vincristine, and mercaptopurine. He developed psoriasis and Crohn’s disease, respectively, 8 and 11 years after end of treatment for the T-ALL.
At time of T-PLL diagnosis, initial workup revealed splenomegaly 7.0 cm below the costal margin, lactate dehydrogenase 865 IU per liter (L), hemoglobin 9.83 mmol/L, platelet count 105 × 109/L, and white blood cell count 483 × 109/L. Blood smear showed small to medium-sized mature lymphocytes with small round to slightly irregular nuclei, some with small nucleoli. The cytoplasm was basophilic, without granulation while a proportion of the cells showed small membranous protrusions. Bone marrow (BM) biopsy demonstrated moderate hypercellularity with infiltration of small lymphocytes, which were immunohistochemically positive for CD3, CD5, CD7 with variable expression of CD8. Negativity for CD2, CD4, TCL-1, CD30, CD56, cytotoxic markers, CD34, TdT and CD1a confirmed the presence of mature lymphocytes. Thus, in conjunction with the morphology of the peripheral blood (PB) lymphocytes, the diagnosis of T-PLL was established ().
Figure 1. (A–E) Bone marrow, year 2000: T-ALL; (A, B) crista biopsy, showing hypercellular BM, with tightly packed blastic cells with large nuclei; (C–E) blasts were positive in CD3 (C), CD5 (D) and CD34 (E). (F–J) Biopsy from colon, year 2013: Indolent T-cell lymphoproliferative disorder of the GI-tract; (F–G) mucosal and submucosal infiltration of small mature looking lymphocytes without evident atypia, deteriorating normal architecture; (H–J) the lymphocytes were positive in CD3 (H), but negative in CD5 (I) and CD34 (J). (K–O) Bone-marrow, year 2017: T-PLL; (K) blood smear with lymphocytosis composed of small to medium-sized mature lymphocytes with small round to slightly irregular nuclei; (L) hypercellular bone-marrow with tightly packed small mature lymphocytes; (M–O) the lymphocytes were positive in CD3 (M) and CD5 (N), negative in CD34 (O). (P) Patient’s leukocyte count was monitored during all treatment regimens. (Q) The bars represent each treatment regimens and the length of it. (R) An overview of the discussed treatment strategies in relation to the mechanism of action. Alemtuzumab has been suggested to induce complement-dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC) and apoptosis. The selective BCL-2 inhibitor, venetoclax, also targets the apoptotic machinery. The well-characterized purine analog, cladribine, inhibits DNA synthesis and has shown to upregulate CD30 expression providing a potential rational for brentuximab vedotin treatment. Another approach suggested in T-PLL has been the use of HDAC inhibitors, e.g. vorinostat. These drugs inhibit HDAC enzymes resulting in increased acetylation of lysine residues on both histone proteins as well as other proteins. Last, based on a hallmark of T-PLL being JAK/STAT genetic alterations, JAK/STAT inhibitors represent another potential strategy.
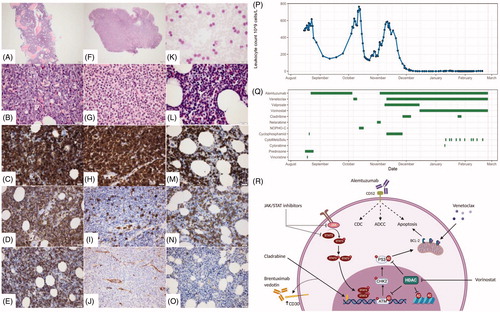
As the diagnosis of T-PLL was confirmed, the treatment strategy was alemtuzumab (30 mg iv three times weekly after initial ramp up), but after 5 weeks of treatment with a nadir lymphocyte count of 160 × 109/L, the patient progressed with lymphocytosis in PB (>600 × 109/L). At progression, the patient received venetoclax based on two recent reports [Citation4,Citation5]. However, despite daily venetoclax ramp up the patient progressed. Next, the patient was switched to high-dose ARA-C and fludarabine combination therapy according to NOPHO high-risk block C treatment for ALL. Again, the disease progressed after a very temporary reduction in the lymphocyte count ().
Ultimately, a combination therapy of cladribine (5 mg per square meter subcutaneous thrice weekly for 5 days every 4 weeks as hypomethylating agent), valproate (900 mg thrice daily) switched to vorinostat (400 mg daily as HDAC inhibitor) in combination with venetoclax (daily ramp up to 400 mg, continuous treatment) and alemtuzumab 30 mg iv 3 times weekly was given [Citation6,Citation7]. At initiation of this combination therapy, the PB lymphocyte count was 600 × 109/L and 77% of the mononuclear cells in the BM were T-PLL cells. Three weeks later, the PB lymphocyte count was 70 × 109/L with 17% BM mononuclear cells being T-PLL; meeting the criteria for partial remission [Citation8]. The T-PLL cells still did not express CD30. Five weeks later just before planned allogeneic transplant, the BM contained less than 1% T-PLL cells. During this successful treatment, the patient developed neutropenia and one episode of fewer without any known microbial focus. Neutropenia was treated with filgrastim, and following the febrile episode, the patient received antifungal treatment throughout the disease course. Unfortunately, a few weeks before the planned transplantation, the patient demonstrated a CNS relapse with cerebrospinal fluid revealing multiple abnormal T-PLL cell and the patient passed away soon thereafter.
To learn from this rare case, we evaluated specimens from the different diseases (T-ALL, Crohn’s disease and T-PLL) by immunohistochemistry and whole exome sequencing.
First, the BM biopsies from the time of diagnosis of T-ALL were reevaluated, thereby confirming the diagnosis of T-ALL ruling out an uncommon early T-PLL case with a late relapse. The T-ALL biopsy revealed extensive infiltration of blasts immunohistochemically ().
Second, the intestinal biopsies diagnosed as Crohn’s disease 4 years prior to T-PLL were reassessed. All seven biopsies from various localizations in the colon showed mucosal and submucosal infiltration of small mature T-lymphocytes without evident atypia, in some areas with crypt abscesses, small reactive follicles, and epithelioid granulomas. The T-cells were immunohistochemically positive for CD2, CD3 and CD7, but negative for CD5, CD4 and CD8. The aberrant phenotype of the T-cells, with loss of CD5, and double negativity for CD4 and CD8 suggests that the T-cells were clonal and that the correct diagnosis is ‘Indolent T-cell lymphoproliferative disorder of the GI-tract’ (GI-T-lymphoma). Clinically and morphologically this can mimic Crohn’s disease [Citation9].
Somatic variants were identified in the T-ALL BM, the GI-T-lymphoma and the T-PLL BM (). Three variants in the T-ALL sample (STAT5B p.Asn642His, JAK3 p.Arg657Gln, and EZH2 p.Ser695Leu) were also identified in the T-PLL sample along with two additional variants in TP53 (p.Phe270Ser and p.Gly246Ser) with different variant allele frequencies (VAF) between the samples (). While the EZH2 variant was the predominant somatic variant in the T-ALL sample (VAF 74%) compared to 17 and 30% for the STAT5B and JAK3 variants, respectively, the T-PLL displayed high frequencies of both STAT5B p.Asn642His and EZH2 p.Ser695Leu (both having VAF of ∼85%) while JAK3 remained at a VAF of 43%. The high VAFs indicate that copy number alterations, such as loss-of-heterozygosity (LOH), may have occurred in these regions. EZH2 is located at chromosome 7q36.1 while STAT5B is located at 17q21.2; SNP array analysis showed LOH at both regions in the T-PLL sample.
Figure 2. Comparison of somatic mutations across different biopsies from the patient. (A) Graphic overview of selected shared and private mutations in the different biological samples at the time point of emergence of the mutations. (B) Variant allele frequencies of the mutations are shown for each biopsy. 1JAK3 mutation was identified in two reads (sequencing depth 105-fold). 2NRAS mutation was only identified in one out of three biopsies. (C) The number of shared SNVs between the samples and a maximum-likelihood phylogenetic tree based on the identified SNVs. Numbers at tree branches are percentage support (0–100%) from 500 bootstrap replicates.
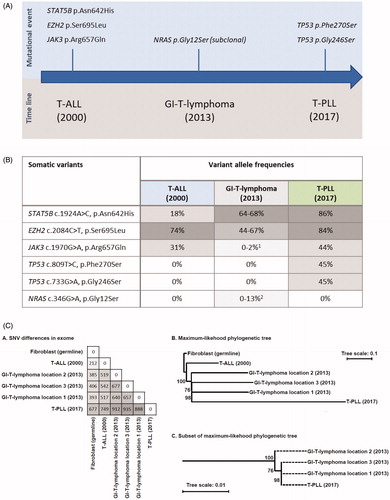
In all three spatially disperse biopsies from the GI-T-lymphoma, STAT5B and the EZH2 variant were identified, indicating that these lesions were indeed related to the diagnosed T-PLL and T-ALL. To explore this further, we performed a phylogenetic analysis using all somatic variants identified in the three tumor samples. We identified 212, 385-406, and 677 single nucleotide variants (SNVs) in the exomes of T-ALL, GI-T-lymphoma and T-PLL samples relative to the fibroblast sample, respectively, and 22 of the variants were found in all five tumor exomes. Only SNV positions sequenced to at least 10-fold coverage depth in all samples were included in the analysis. T-PLL and GI-T-lymphoma samples were most closely related, with a more distant relation to the initial T-ALL () The lack of the JAK3 variant in two out of three biopsies and the different genetic pattern illustrates the heterogenous nature of the cancer cells.
We detected germline variants that might contribute to the development of T-cell malignancies. Two heterozygous, likely pathogenic germline variants in DNA repair genes were identified; (1) FANCC (NM_000136.2), c.705del, p.Met236Cysfs*4 and (2) BLM (NM_001287246.1), c.2488dup, p.Thr830Asnfs*5. The FANCC variant has not been reported in the Genome Aggregation Database (gnomAD) version 2.0 or in the literature, while other variants in FANCC has been found in patients with Fanconi anemia in a compound heterozygous or homozygous state.17 The BLM variant has been reported in gnomAD with an allele frequency of 0.0012% and is associated with Blooms syndrome in compound heterozygous or homozygous state [Citation10,Citation11].
Discussion
Here we report a rare case of alemtuzumab-refractory T-PLL in a 26-year-old male with an unusual prior history of T-ALL, psoriasis and GI-T-lymphoma initially misdiagnosed as Crohn’s disease. Alemtuzumab combined with conventional chemotherapy has not shown to be superior to single-agent alemtuzumab, therefore the treatment regimen was based on case reports and translational findings on the use of novel-targeted agents in T-PLL and very limited data on treatment of alemtuzumab refractory T-PLL [Citation6,Citation7,Citation12,Citation13]. An overview of the therapeutic options discussed is shown in .
The Arg642His STAT5B variant has been identified in several types of aggressive T-cell lymphomas, and is described as a gain-of-function variant [Citation14]. The JAK3 variant is also described as a gain-of-function variant that is often present in T-PLL and may pose a target for novel treatment approaches [Citation15]. Retrospectively, these STAT5B and JAK3 recurrent mutations could have provided a rationale for adding a JAK/STAT inhibitor e.g. the JAK3 inhibitor tofacitinib, which might have deepened the response further and prevented the CNS relapse as tofacinib may pass the blood–brain barrier [Citation15,Citation16]. Additionally, we considered to add brentuximab vedotin based on prior reports on increased CD30 expression upon cladribine treatment in T-PLL, but refrained from this due to the absence of CD30 positive cells in the samples from this patient [Citation7].
On the assumption that sequencing will be part of standard diagnostic work-up in the near future, the many shared mutations would have revealed the indolent GI lymphoma. An initial correct and early diagnosis of the GI-T lymphoma might have benefited the patient, as the T-PLL might have been treated at an earlier state or allogeneic transplant might have been chosen early on as a curative treatment.
In summary, we describe a rare case with multiple T-cell malignancies dispersed over a 16-year period with genetically linked clones, indicating a common dormant leukemic stem cell as well as potential germline mutations predisposing to T-cell malignancies. The described germline variants reveal the complexity and current inconclusive results from the assessment of heterozygous germline variants and their impact on the risk of specific malignancies and autoimmune disease states. Based on large scale genomic projects around the world, this may change significantly over the coming years. The genetic analyses performed retrospectively of different specimens from this patient reveals that broad sequencing not only can confer prognostic information for individual tumors, but also help unravel differential diagnoses between inflammatory and malignant diseases and point toward treatment options. The partly successful novel treatment approach presented here with a combination of alemtuzumab, venetoclax, vorinostat and cladribine emphasizes the importance of knowledge accumulation of treatment approaches and their outcome in rare diseases.
Disclosure statement
RV has received travel grants from Roche and Abbvie. CUN has received consultancy fees and/or travel grants from Janssen, Abbvie, Novartis, Roche, Sunesis, Gilead, AstraZeneca, and CSL Behring, research support from Abbvie, AstraZeneca and Janssen, outside this project. KS has received speaker and/or advisory Board Honoraria from Amgen, Jazz Pharmaceuticals, Servier, and Medscape and educational grant from Servier. MAA, LDS, LB, MKA, RLM and CWY declare they have nothing to disclose.
Additional information
Funding
References
- Herling M, Khoury JD, Washington LBT, et al. A systematic approach to diagnosis of mature T-cell leukemias reveals heterogeneity among WHO categories. Blood. 2004;104(2):328–335.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390.
- Taylor A, Metcalfe J, Thick J, et al. Leukemia and lymphoma in ataxia telangiectasia. Blood. 1996;87(2):423–438.
- Andersson EI, Pützer S, Yadav B, et al. Discovery of novel drug sensitivities in T-PLL by high-throughput ex vivo drug testing and mutation profiling. Leukemia. 2018;32(3):774–787.
- Boidol B, Kornauth C, Van Der Kouwe E, et al. First-in-human response of BCL-2 inhibitor venetoclax in T-cell prolymphocytic leukemia. Blood. 2017;130(23):2499–2503.
- He L, Tang J, Andersson EI, et al. Patient-customized drug combination prediction and testing for t-cell prolymphocytic leukemia patients. Cancer Res. 2018;78(9):2407–2418.
- Hasanali ZS, Saroya BS, Stuart A, et al. Epigenetic therapy overcomes treatment resistance in T cell prolymphocytic leukemia. Sci Transl Med. 2015;7(293):293ra102–293ra102. ra
- Staber PB, Herling M, Bellido M, et al. Consensus criteria for diagnosis, staging, and treatment response assessment of T-cell prolymphocytic leukemia. Blood. 2019;134(14):1132–1143.
- Soderquist CR, Bhagat G. Gastrointestinal T- and NK-cell lymphomas and indolent lymphoproliferative disorders. Semin Diagn Pathol. 2020;37(1):11–23.
- Schoenaker MHD, Henriet SS, Zonderland J, et al. Immunodeficiency in Bloom's syndrome. J Clin Immunol. 2018;38(1):35–44.
- German J, Sanz MM, Ciocci S, et al. Syndrome-causing mutations of the BLM gene in persons in the Bloom's Syndrome Registry. Hum Mutat. 2007;28(8):743–753.
- Sud A, Thomsen H, Sundquist K, et al. Risk of second cancer in Hodgkin lymphoma survivors and influence of family history. J Cin Oncol. 2017;35(14):1584–1590.
- Dearden C. Disease-specific complications of chronic lymphocytic leukemia. Hematol Am Soc Hematol Educ Progr. 2008;2008(1):450–456.
- Springuel L, Renauld JC, Knoops L. JAK kinase targeting in hematologic malignancies: a sinuous pathway from identification of genetic alterations towards clinical indications. Haematologica. 2015;100(10):1240–1253.
- Li G, Waite E, Wolfson J. T-cell prolymphocytic leukemia in an adolescent with ataxia-telangiectasia: novel approach with a JAK3 inhibitor (tofacitinib). Blood Adv. 2017;1(27):2724–2728.
- Fukuyama T, Tschernig T, Qi Y, et al. Aggression behaviour induced by oral administration of the Janus-kinase inhibitor tofacitinib, but not oclacitinib, under stressful conditions. Eur J Pharmacol. 2015;764:278–282.