
Background
The BRCA1-associated protein-1 (BAP1) gene has in the recent years been identified as a critical driver gene in the pathogenesis of many tumors [Citation1,Citation2]. The BAP1 gene is located on chromosome 3 (3p21.31-p21.2), where its 17 exons transcribe into the 729 amino acids long BAP1 protein [Citation3]. BAP1 has been categorized as a tumor suppressor, and has roles in numerous cellular processes, including DNA damage response, cell cycle regulation, cell growth, metabolism, and the regulation of inflammatory responses [Citation1,Citation3,Citation4]. It is known to bind to a number of proteins via specific domains, including BRCA1, BARD1, ASXL1/2, HCFC1, YY1, and FOXK1/2 (11) as indicated in [Citation3,Citation5,Citation6]. BAP1 is frequently mutated or lost in several tumor types, including uveal melanoma (UM), cholangiocarcinoma, renal cell carcinoma (RCC), mesothelioma, and bladder cancer [Citation2,Citation7]. Somatic loss of BAP1 in tumors is also in many tumors associated with a poor outcome [Citation8–10]. The first report of a germline pathogenic variant (PV) in BAP1 was published in 2011 [Citation8]. Subsequent reports described inactivating germline PVs segregating in cancer-prone families, mainly characterized by distinct melanocytic tumors and mesothelioma in combination with other cancers [Citation11–14]. The neoplasms associated with BAP1 germline PVs, also called the BAP1 tumor predisposition syndrome (TPDS), has been defined to include the core tumors cutaneous melanoma (CM), UM, RCC, pleural and peritoneal mesothelioma, meningioma, basal cell carcinoma (BCC), and BAP1-inactivated melanocytic tumor (BIMT) [Citation11,Citation15–17]. BIMTs or informally BAPomas are skin-colored to reddish-brown papules that have characteristic clinical and dermatoscopic features and a unique melanocytic pathology [Citation13,Citation18]. Here, we report of the BAP1 germline genetics testing in melanoma and cancer-prone families in Sweden that has been ongoing since the identification of the first carrier family in 2012.
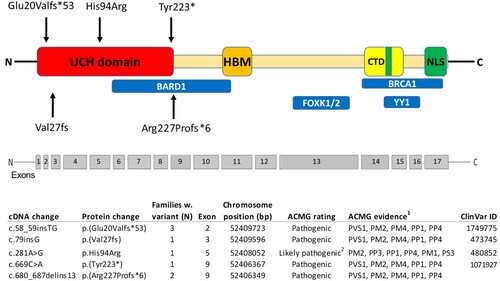
Figure 1. Pathogenic variants identified in Sweden, plotted along the BRCA1-associated protein-1 (BAP1) gene with the functional domains shown. Ubiquitin carboxyl hydrolase (UCH) domain; HBM, host cell factor 1 (HCF1) binding domain; nuclear localization signals (NLS); C-terminal domain (CTD), additional sex combs like (ASXL1/2) binding domain; BRCA1-associated RING domain protein 1 (BARD1) binding region; Breast Cancer type 1 (BRCA1)-binding region and Ying Yang 1 (YY1) binding region; Forkhead Box Protein K1/2. 1American College of Medical Genetics (ACMG) evidence: PVS1 - null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multi-exon deletion) in a gene where LOF is a known mechanism of disease. PM1-mutational hotspot, PM2–Absent from controls in Exome Aggregation Consortium (ExAC). PM4–Protein length changes as a result of in-frame deletions/insertions in a nonrepeat region or stop-loss variants. PP1–Cosegregation with disease in multiple affected family members in a gene definitively known to cause the disease. PP3–Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.). PP4–Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology. PS3–Functional studies. 2Functional studies of the missense variant c.281A > G, demonstrated an almost complete abolishment of enzymatic activity (Repo et al. Hum Mol Genet. 2019). This together with a co-segregation of a highly specific phenotype, absence of variant in controls, computational evidence and position in a hotspot gene site gives evidence that the variant is likely pathogenic.
Material and methods
Patients and families
After the identification of the first family in Sweden with a BAP1 PV in 2012, germline sequencing of the BAP1 gene has been offered to families presenting with BAP1 core tumors. Essentially, this has been performed in families with CM in combination with any other BAP1 core tumor in the first- or the second-degree relatives (UM, BIMT, mesothelioma, meningioma, or RCC). Also, families with two or more cases of UM or UM in combination with any other BAP1 core tumor in the first- to the second-degree relatives have been tested. Similarly, families with BIMTs in combination with any other BAP1 core tumor has been tested. Families with suspected BAP1 tumor syndrome were on a national level, referred to the Hereditary melanoma clinic at Karolinska University hospital in Stockholm, where the test has been performed. In addition, since 2019, all ‘regular’ CM families in Stockholm (i.e., without BAP1 core tumors) have undergone BAP1 testing as it has been implemented in routine gene panel testing for familial melanoma (including also CDKN2A and CDK4). For this testing, familial melanoma has been defined as three or more cases of CM in a family or two CM cases if one case is <55 years at diagnosis. At the hereditary melanoma clinic, a pedigree has been established and diagnoses verified by pathology or clinical records and if the criteria for genetic testing is fulfilled, a test has been offered to affected members. Descriptive statistics were applied to demonstrate the occurrence of BAP1 PVs in association with tumor spectrums in the families. The study was approved by the Swedish Ethical Review Authority (Dnr. 2012/1192-32 and 2018/803-31).
BAP1 germline genetic testing
Until December 2018, genomic DNA was analyzed for genetic variation in the BAP1 gene in a research laboratory with Sanger sequencing (bidirectional). All exons and exon–intron boundaries were covered by the analysis. As of January 2019, the screening was done as a clinical test at an accredited laboratory, the Karolinska University Laboratory. The method used was accredited with a library preparation performed with IonAmpliSeq sequencing with IonS5 and data analysis with IonReporter (Life Technologies, Carlsbad, CA). All the reported gene variants have been detected by both methods. The clinical variant interpretation guidelines from the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) were used to assess if a variant was (likely) pathogenic, of uncertain significance, or (likely) benign [Citation19].
Results
Incidence of BAP1 PVs in tested families
In total, 190 families were tested for BAP1 PVs. Of these, 141 were CM families where none of the other BAP1 core tumors (UM, BIMT, RCC, mesothelioma, or meningioma) have been identified (). In these families none (0%) were identified with a BAP1 PV. Among these 141 CM families, none (0%) had CDK4 PVs while nine (6.4%) had CDKN2A PVs (same PVs as previously reported) [Citation20]. There were 49 families that were tested specifically for a suspicion of BAP1 tumor syndrome (). BAP1 PVs were identified in eight (16.3%) of these families. Families with CM, UM, or RCC in family all had similar frequencies of BAP1 PV (<20%). BIMTs were diagnosed in nine families of which seven (77.8%) carried a germline BAP1 PV. Mesotheliomas and meningiomas were diagnosed in six and four families of which five (83.3%) and two (50%) had BAP1 PVs, respectively. In families with BIMT in combination with another BAP1 core tumor type in family, the frequency of BAP1 PVs was high (>85%).
Table 1. BAP1 germline genetic testing in 190 families from Sweden.
PVs and tumor spectrum in BAP1 carrier families
Eight families with BAP1 PVs were identified, of which four have been previously reported [Citation17,Citation21] (). Of the eight families, seven (87.5%) families had CM in their pedigree, five (62.5%) had UM, seven (87.5%) had BIMT, five (62.5%) had mesothelioma, two (25.0%) had meningioma, and two (25.0%) had RCC, while six families (75.0%) had BCC in their pedigree. The youngest age at diagnosis in the families was, for CM, median 44.5 years (range 20–56 years), for UM 36 years (range 16–40 years), for BIMT 11 years (range 10–51 years), for mesothelioma 52 years (range 39–80 years), for meningioma 50.5 years (range 40–61 years), for RCC 67.5 years (range 66–69 years), and for BCC 52 years (range 39–76 years). Among families with BAP1 core tumors, tested negative for BAP1 PV, the median age at diagnosis of CM was 45 years (range 16–80 years) and for UM 58 years (range 20–72 years). Hence UM were diagnosed in significantly younger members in BAP1 PV families (p < .001), whereas CM were diagnosed at similar ages in carrier and noncarrier families (p = .594). Other tumors that were present in the pedigrees of at least two families were breast, pancreatic, prostate, and lung cancer. Highest mortality was seen for mesothelioma, where six of seven cases (83.3%) had died from the disease, the only survivor was diagnosed with a lesion considered to be a mesothelioma precursor. Of the eight cases of UM, four had died from the disease (50%), whereof three deaths were in the same family. Among the 12 cases of CM, only one died from CM (8.3%). Both cases of RCC died from their disease.
Table 2. Tumors in BAP1 pathogenic variant carrying families identified in Sweden.
Among the eight families, five different BAP1 PVs were identified (), where none has been reported in the general population. Interestingly, all the PVs identified in the Swedish families are located in the first part of the gene, four of them are null variants, predicted to lead to a truncated protein lacking several important binding domains downstream or to activate nonsense-mediated RNA decay (NMD) of the aberrant transcripts (). The fifth variant (His94Arg) is a missense variant located in the ubiquitin carboxy-terminal hydrolase (UCH)-domain. In a comprehensive study including known PVs (Walpole et al.2018), all of the BAP1 missense variants classified as likely pathogenic were actually located in the UCH-domain [Citation17]. All but one PV, c.669C > A (p.(Tyr223*), have been previously reported [Citation17,Citation21]. The Tyr223* variant is considered pathogenic and was identified in a proband that at an age of 46 had been diagnosed with several tumors, including CM, BIMT, and bilateral breast cancer. In the relatives, several other tumors had also been diagnosed (). This PV is a nonsense variant introducing a premature termination codon in exon 9 of the BAP1 gene.
Three families (of which one has been previously reported [Citation17]) have been identified with the same PV, c.58_59insTG (p(Glu20Valfs*53)), indicating a founder effect in Sweden for this variant that has, to our knowledge, not been identified in other countries. In the families with this PV, all the BAP1 core tumors, beside RCC have been diagnosed (). This PV is located in exon 2 and is a 2-base-pair insertion causing a shift in the reading frame and subsequent introduction of a premature termination codon 53 amino acid downstream of the insertion.
Two families (of which one has been previously reported [Citation17]) have been identified with the same PV, c.680_687delins13 (p.(Arg227Profs*6)). Also, this variant has not been reported outside Sweden. In these two families, all the BAP1 core tumors, beside meningioma, have been diagnosed (). This pathogenic frame-shift variant is also located in exon 9 and leads to a truncated protein or to NMD.
Further, two families with different variants, c.79insG (p.(Val27fs)) and c.281A > G, (p.(His94Arg)), have been previously reported where the former has only been detected in Sweden and the latter is in a family with members living in both Sweden and Finland [Citation17,Citation21,Citation22]. The first is a null variant, caused by a 1-base-pair insertion in exon 3, causing a frameshift and a premature stop codon. The latter is a missense variant in exon 5 that is considered as likely pathogenic. It is located in the middle of the UCH-domain and functional studies in vitro shows that the His94Arg variant almost entirely abolish the deubiquitinating activity [Citation22]. In both families, several BAP1 core tumors have been diagnosed ().
The segregation patterns in the BAP1 families are presented in Supplementary Table S1. To summarize, for each of the PVs, several individuals with BAP1 core tumors have been tested positive for BAP1 PV (2–10 members for each of the PVs). In none of the BAP1 PV families, there has been a ‘phenocopy’, e.g., a BAP1 wt member with any of the BAP1 core tumors (uveal melanoma, cutaneous melanoma, BIMT, mesothelioma, meningioma, or renal cell cancer). A lesser number of individuals without BAP1 core tumors has been available for BAP1 testing (1–6 members for each of the PVs). The members tested as carriers that have no diagnoses of any BAP1 core tumor are all still at a rather young age (<55 years). Another aspect is that germline missense BAP1 variants have recently been associated with a rare syndromic neurodevelopmental disorder (NDD) [Citation23]. In the identified BAP1 families in Sweden, there is no known case of NDD.
Discussion
To summarize, our study shows that in CM families lacking the other typical BAP1 core tumors, BAP1 PV variants are extremely uncommon (0% of the Swedish families). This is in line with findings from a Dutch study where BAP1 PVs were found in 0.7% of CM families, however, two of the identified families also had BIMTs [Citation24]. Further, in a large English cohort of CM families, deleterious BAP1 variants were found in 0.2% of families, but also here, the pedigrees of carrier families revealed other BAP1 core tumors [Citation25]. Similar findings have been reported in samples of CM families from Denmark, Australia, and USA, with BAP1 PVs present in well below 1% of CM families [Citation15,Citation16,Citation26]. In a study from Finland, BAP1 PVs were found in 25% of UM families. Further, in rare families with multiple cases of mesothelioma, BAP1 PVs are prevalent [Citation12,Citation27,Citation28]. In the Swedish families included, presenting with different BAP1 core tumors, a significant portion (16%) was identified with BAP1 PVs. In fact, in families having BIMT together with any other core tumor, BAP1 germline mutations are so prevalent (>85% of the Swedish families), that in such families, BAP1-TPDS should always be suspected. Similarly, families presenting with both UM and mesothelioma that are both uncommon tumors in the normal population should always raise suspicion of BAP1-TPDS. Of note, neither the Sanger sequencing nor the IonAmpliSeq method used in our study has the potential to detect structural rearrangements or copy number changes (larger deletions or duplications). Hence, it is likely that if such a family exists in our cohort, it would have been missed. Still there have been very few families detected with such large germline BAP1 aberrations, to our knowledge only one family with a large deletion [Citation17,Citation29].
The strength of the study is the population-based setting, with testing on a national level for the BAP1 mutation. A possible bias is that our clinical and research focus is on melanocytic tumors and skin tumors (CM, UM, BIMT, BCC), hence, potential families presenting with only the other BAP1 core tumors (mesothelioma, RCC, or meningioma), could therefore have been missed. The tumors that are typical for the BAP1-TPDS are usually diagnosed and followed-up by different healthcare specialists (dermatologists, ophthalmologists, lung specialists, urologists, neurologists, oncologists etc.). This study hopefully contributes to an increased awareness among medical professionals that encounter patients with personal and family history of these specific tumors that should be referred for genetic testing. Identification of families with a germline BAP1 PV is essential for early detection and appropriate surveillance for individuals that are at high risk for a broad spectrum of tumors. Our study supports the initiation of dermatologic follow-up of BAP1 carriers in early adolescence because skin tumors (CM, BIMT and BCC) are invariably seen in the families and can present at a young age. Similarly, to initiate ophthalmologic controls in early adulthood (or in adolescence in families with very young case(s)) appears rational, considering the early onset of UM that was seen in some of the carriers. In our study, RCC was only seen among two carriers of one of the specific PVs, Arg227Profs*6 both in their late sixties, but were advanced tumors since the patients died from their disease. In the comprehensive study from 2018, lower incidence of RCC was also seen, compared to CM, UM, and mesothelioma [Citation17]. With the current knowledge we argue toward that RCC screening (ultrasound or MRI) should be directed toward those with a family history of RCC or PVs known to be associated with this tumor, starting at age 40, or 10 years earlier than the earliest case in the family. There are to date no screening modalities that have been established in the screening for mesothelioma [Citation30]. However, due to the high incidence of different tumors (also other than the core tumors), there should be a low threshold to initiate directed investigations (radiology, endoscopy, pathology etc.) in carriers presenting with different symptoms. To conclude, in Sweden, BAP1 mutations are found in families presenting with specific tumors where members require directed counseling and surveillance. This study has further contributed to the setting of the national recommendations for genetic testing and surveillance of BAP1 families in Sweden [Citation31].
Supplemental Material
Download MS Word (14.5 KB)Disclosure statement
No potential conflict of interest was reported by the authors
Data availability statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Additional information
Funding
References
- Eletr ZM, Wilkinson KD. An emerging model for BAP1's role in regulating cell cycle progression. Cell Biochem Biophys. 2011;60(1-2):3–11.
- Helgadottir H, Hoiom V. The genetics of uveal melanoma: current insights. Appl Clin Genet. 2016;9:147–155.
- Jensen DE, Proctor M, Marquis ST, et al. BAP1: a novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene. 1998;16(9):1097–1112.
- Yu H, Pak H, Hammond-Martel I, et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci USA. 2014;111(1):285–290.
- Yu H, Mashtalir N, Daou S, et al. The ubiquitin carboxyl hydrolase BAP1 forms a ternary complex with YY1 and HCF-1 and is a critical regulator of gene expression. Mol Cell Biol. 2010;30(21):5071–5085.
- Ji Z, Mohammed H, Webber A, et al. The forkhead transcription factor FOXK2 acts as a chromatin targeting factor for the BAP1-containing histone deubiquitinase complex. Nucleic Acids Res. 2014;42(10):6232–6242.
- Dono M, Angelini G, Cecconi M, et al. Mutation frequencies of GNAQ, GNA11, BAP1, SF3B1, EIF1AX and TERT in uveal melanoma: detection of an activating mutation in the TERT gene promoter in a single case of uveal melanoma. Br J Cancer. 2014;110(4):1058–1065.
- Harbour JW, Onken MD, Roberson ED, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330(6009):1410–1413.
- van Essen TH, van Pelt SI, Versluis M, et al. Prognostic parameters in uveal melanoma and their association with BAP1 expression. Br J Ophthalmol. 2014;98(12):1738–1743.
- Luchini C, Veronese N, Yachida S, et al. Different prognostic roles of tumor suppressor gene BAP1 in cancer: a systematic review with meta-analysis. Genes Chromosom Cancer. 2016;55(10):741–749.
- Abdel-Rahman MH, Pilarski R, Cebulla CM, et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011;48(12):856–859.
- Testa JR, Cheung M, Pei J, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43(10):1022–1025.
- Wiesner T, Obenauf AC, Murali R, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43(10):1018–1021.
- Aoude LG, Wadt K, Bojesen A, et al. A BAP1 mutation in a Danish family predisposes to uveal melanoma and other cancers. PLoS One. 2013;8(8):e72144.
- Njauw CN, Kim I, Piris A, et al. Germline BAP1 inactivation is preferentially associated with metastatic ocular melanoma and cutaneous-ocular melanoma families. PLoS One. 2012;7(4):e35295.
- Wadt KA, Aoude LG, Krogh L, et al. Molecular characterization of melanoma cases in Denmark suspected of genetic predisposition. PLoS One. 2015;10(3):e0122662.
- Walpole S, Pritchard AL, Cebulla CM, et al. Comprehensive study of the clinical phenotype of germline BAP1 variant-carrying families worldwide. J Natl Cancer Inst. 2018;110(12):1328–1341.
- Yelamos O, Navarrete-Dechent C, Marchetti MA, et al. Clinical and dermoscopic features of cutaneous BAP1-inactivated melanocytic tumors: results of a multicenter case-control study by the international dermoscopy society. J Am Acad Dermatol. 2019;80(6):1585–1593.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424.
- Pissa M, Helkkula T, Appelqvist F, et al. CDKN2A genetic testing in melanoma-prone families in Sweden in the years 2015-2020: implications for novel national recommendations. Acta Oncol. 2021;60(7):888–896.
- Hoiom V, Edsgard D, Helgadottir H, et al. Hereditary uveal melanoma: a report of a germline mutation in BAP1. Genes Chromosom Cancer. 2013;52(4):378–384.
- Repo P, Jarvinen RS, Jantti JE, et al. Population-based analysis of BAP1 germline variations in patients with uveal melanoma. Hum Mol Genet. 2019;28(14):2415–2426.
- Kury S, Ebstein F, Molle A, et al. Rare germline heterozygous missense variants in BRCA1-associated protein 1, BAP1, cause a syndromic neurodevelopmental disorder. Am J Hum Genet. 2022;109(2):361–372.
- Potjer TP, Bollen S, Grimbergen A, et al. Multigene panel sequencing of established and candidate melanoma susceptibility genes in a large cohort of Dutch non-CDKN2A/CDK4 melanoma families. Int J Cancer. 2019;144(10):2453–2464.
- O'Shea SJ, Robles-Espinoza CD, McLellan L, et al. A population-based analysis of germline BAP1 mutations in melanoma. Hum Mol Genet. 2017;26(4):717–728.
- Aoude LG, Gartside M, Johansson P, et al. Prevalence of germline BAP1, CDKN2A, and CDK4 mutations in an Australian Population-Based sample of cutaneous melanoma cases. Twin Res Hum Genet. 2015;18(2):126–133.
- Ohar JA, Cheung M, Talarchek J, et al. Germline BAP1 mutational landscape of asbestos-exposed malignant mesothelioma patients with family history of cancer. Cancer Res. 2016;76(2):206–215.
- Repo P, Staskiewicz A, Sutinen E, et al. BAP1 germline variants in Finnish patients with malignant mesothelioma. Lung Cancer. 2022;165:102–107.
- Pandithan D, Klebe S, McKavanagh G, et al. BAP1 tumour predisposition syndrome due to whole BAP1 gene deletion. Case Rep Genet. 2022;2022:5503505.
- van Meerbeeck JP, Hillerdal G. Screening for mesothelioma: more harm than good? Am J Respir Crit Care Med. 2008;178(8):781–782.
- Swedish national guidlines familial melanoma. RCC; 2022. https://kunskapsbanken.cancercentrum.se/globalassets/cancerdiagnoser/hud/vardprogram/hela-bilaga-1.-melanom-familjara-melanom.pdf