
Abstract
Background: The introduction of new disease-modifying therapies (DMTs) for remitting-relapsing multiple sclerosis (RRMS) has considerably transformed the landscape of therapeutic opportunities for this chronic disabling disease. Unlike injectable drugs, oral DMTs promote patient satisfaction and increase therapeutic adherence.
Review: This article reviews the salient features about the mode of action, efficacy, safety, and tolerability profile of approved oral DMTs in RRMS, and reviews their place in clinical algorithms in the Middle East and North Africa (MENA) region. A systematic review was conducted using a comprehensive search of MEDLINE, PubMed, Cochrane Database of Systematic Reviews (period January 1, 1995–January 31, 2018). Additional searches of the American Academy of Neurology and European Committee for Treatment and Research in Multiple Sclerosis abstracts from 2012–2017 were performed, in addition to searches of the Food and Drug Administration and European Medicines Agency websites, to obtain relevant safety information on these DMTs.
Conclusions: Four oral DMTs: fingolimod, teriflunomide, dimethyl fumarate, and cladribine have been approved by the regulatory agencies. Based on the number needed to treat (NNT), the potential role of these DMTs in the management of active and highly active or rapidly evolving RRMS is assessed. Finally, the place of the oral DMTs in clinical algorithms in the MENA region is reviewed.
Introduction
Since the introduction of interferon-β (IFNβ) and glatiramer acetate (GA) more than two decades ago, the rapidly changing and dynamic treatment landscape of multiple sclerosis (MS) has become increasingly challenging for the treating neurologists and patients alike. In addition, it has created opportunities to treat MS at an early stage and with much more aggressive disease-modifying therapies (DMTs) (e.g. alemtuzumab, ocrelizumab), relative to the past. In September 2010, the first oral DMT, fingolimod, was approved by the Food and Drug Administration (FDA) for patients with remitting relapsing multiple sclerosis (RRMS). While many of the newly introduced DMTs for RRMS are subject to certain safety issues, the concern with injectable DMTs (IFNβ and GA) was more focused on tolerability and adherence. The proportion of patients adherent to injectable DMTs varied between 55–90%Citation1,Citation2, and persistence after 1 year was ∼50%Citation3. A systematic review by Giovannoni et al.Citation4 on observational and randomized controlled trials of injectable DMTs revealed that the most-cited reasons for discontinuation of therapy were adverse events and lack of efficacy. In addition, this review also revealed a significant incidence of flu-like symptoms and injection site reactions that persisted over time. These tolerability issues proved to have a direct impact on persistence and adherenceCitation5,Citation6.
As of 2017, a fourth oral DMT (cladribine) has become available for RRMS as an alternative to injectable DMTs. Randomized, double-blind, placebo-controlled trials in RRMS revealed that the oral DMTs (fingolimod, teriflunomide, dimethyl fumarate, and cladribine) significantly reduced the annualized relapse rate (ARR) and the occurrence of MRI activity when compared with placeboCitation7–9. The four oral DMTs differ in their dosing regimen. Fingolimod (0.5 mg) and teriflunomide (14 mg) are taken once daily, and neither have an initiation dose in their regimensCitation10,Citation11. In contrast, the initial dose of dimethyl fumarate is 120 mg BID for 7 days followed by a maintenance dose of 240 mg BIDCitation12. Patients who do not tolerate the dimethyl fumarate maintenance dose should be subjected to temporary dose reduction to 120 mg BID, but the full maintenance dose should be resumed within 4 weeks. Only patients taking fingolimod are required to have a minimum 6 h first dose observation with cardiac monitoring. Cladribine is given as a short-course, annual oral dosing regimen. Each treatment course consists of two treatment weeks (one at the beginning of the first month and one at the beginning of the second month). Each treatment week consists of 4 or 5 days on which a patient receives 10 mg or 20 mg (one or two tablets) as a single daily dose, depending on the patient’s body weight. This course is given for two consecutive years. Following completion of the second treatment courses, no further cladribine treatment is required in years 3 and 4.
The prevalence of MS varies considerably across different areas of the world. A recent meta-analysis revealed that the overall MS prevalence in the Middle East and North Africa (MENA) region is 51.5/100,000Citation13. In addition, the MS prevalence is also much higher in the Arabian Gulf countries (54.8–85.0/100,000)Citation14–16, with an increasing trend over the past decadesCitation14,Citation17.
In the fast developing economic countries of the Arabian Gulf (Bahrain, Kuwait, Oman, Qatar, Saudi Arabia, United Arab Emirates), the therapeutic armamentarium of RRMS includes all Food and Drug Administration (FDA) and European Medicines Agency (EMA) approved agents, without exception. Unlike in many Western countries, the Arabian Gulf countries have no medical criteria-based healthcare reimbursement system. All governmental health services, including DMTs for RRMS, are free-of-charge for their nationals. Needless to say that, in such a setting, decision-making processes about DMTs may be challenging. Adding to this are the different geographical training background, culture, and personal experience of the practicing neurologists, and, last but not least, the patient’s individual needs and preferences. In other parts of the MENA region the availability of these drugs is subject to regulations of the local health authorities, but few DMTs for RRMS are available.
The objective of this paper is to summarize the salient features on the mode of action, clinical efficacy, safety, and tolerability of the approved oral DMTs in the treatment of RRMS, and review their place in clinical algorithms in the MENA region.
Methods
A systematic literature review was performed through a comprehensive search of MEDLINE, PubMed, Embase, Cochrane Database of Systematic Reviews (period January 1, 1995–January 31, 2018) using a combination of medical subject headings (MeSH) and free-text terms to identify publications of relevant studies of approved oral DMTs for RRMS. The following search terms were used: “multiple sclerosis”, “randomized controlled trials”, “oral disease-modifying therapies”, “teriflunomide”, “fingolimod”, “dimethyl fumarate”, “cladribine”, “progressive multifocal leukoencephalopathy”, “guidelines”, “recommendations”, “algorithm”, and “Middle East and North Africa”. Conference abstracts were included and identified via Embase or web-searches of relevant conference websites. Additional searches were performed of American Academy of Neurology (AAN) and European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) abstracts from 2012–2017 using identical search strategies on their respective websites. In addition, searches of Trial registries (e.g. ClinicalTrials.gov and European Clinical Trial Registry [EUCTR]), FDA, and EMA websites were also performed. Articles and abstracts retrieved needed to be original reports, and of relevance to the scope of this manuscript. Full-text versions of articles were examined. No language restrictions were imposed on the retrieved articles.
Clinical (annualized relapse rate (ARR), relapse freedom, increase in Expanded Disability Status Scale (EDSS) or EDSS progression sustained for 3 or 6 months) and neuroimaging (lack of new/enlarging T2 and new gadolinium-enhancing (Gad+) lesions) outcome measures were used to evaluate efficacy. In addition, brain volume loss (BVL)—assumed to be a surrogate marker of long-term disability—and “No Evidence of Disease Activity (NEDA)” were included. NEDA is defined by three related measures of disease activity: (i) no confirmed relapses (inflammatory disease activity); (ii) no confirmed disability progression as measured on the EDSS, (iii) no MRI activity (new or enlarging T2 lesions or Gad + lesions)Citation18,Citation19. Referring to BVL, the mean annual rate of BVL in patients with MS is ∼0.5–1.35% per year, which is faster than in comparable age-adjusted healthy individuals (<0.4%)Citation20.
This study (MRC-01-18-034) was granted approval by the Medical Research and Ethics Committee of our institution.
Definitions of clinical events and identification of the clinical course of MS
A clinical relapse is defined as new or worsening symptoms that lasted ≥24 h and occurs in the absence of fever or infection, while disability progression is defined as an increase in EDSS of ≥1 point (or ≥0.5 point for baseline EDSS ≥5.5), confirmed during two subsequent neurological examinations separated by an interval of at least 3–6 months free of relapsesCitation21.
Active RRMS is defined as ≥2 clinically significant relapses in the last 2 years, ≥1 disabling relapse in the last year, or an active MRI containing new or Gad + lesions that have developed during the last yearCitation21. Highly active RRMS should have at least one of the following: (1) ≥ 2 relapses in the previous year with ≥1 Gad + lesions on brain MRI or a significant increase in T2 lesion load as compared to a previous recent MRI, or (2) sub-optimal treatment response to an adequate course of ≥1 DMT(s) and presenting with ≥1 relapse in the previous year while on therapy and having ≥9 T2 lesions or ≥1 Gad + lesionCitation22.
Overview of oral disease-modifying therapies
Fingolimod
Fingolimod (0.5 mg once daily) was the first oral DMT approved by the FDA and the EMA for the treatment of RRMSCitation23,Citation24. Fingolimod is phosphorylated by sphingosine kinase to its active form, fingolimod phosphate, and acts as an agonist on the sphingosine-1-phosphate (S1P)-1 sub-type receptorCitation24,Citation25. This S1P-1 receptor is found on the surface of lymphocytes and central nervous system (CNS) cells. The activation of the S1P-1 receptor prevents autoreactive T and B lymphocytes to egress from the lymph nodes, resulting in a reduction of circulating memory T cells of >70%Citation25. This process potentially reduces the infiltration of pathogenic lymphocytes into the CNS and consequently immunosuppression. Moreover, its lipophilic nature allows it to cross the blood–brain barrier and bind to the S1P-1 receptors on CNS cellsCitation26,Citation27.
Fingolimod’s pharmacokinetic properties have been extensively studied in healthy volunteers and MS patientsCitation28. The drug is slowly (Tmax 12–24 h) but efficiently absorbed, with a high oral bioavailability (93%). As its absorption is unaffected by dietary intake, it can be taken regardless of mealsCitation28. Fingolimod and its active metabolite bind extensively to plasma proteins (99.7%) and are largely cleared through a metabolic pathway that predominantly utilizes cytochrome P450 (CYP) 4F2 followed by fatty acid-like degradation to inactive metabolites and formation of inactive ceramide metabolitesCitation28. Owing to a high volume of distribution, fingolimod’s half-life (t½) is 6–9 days, and steady-state pharmacokinetics are reached after 1–2 months of daily dosing. Approximately 81% of the administered dose is eliminated via urine as inactive metabolitesCitation28.
The large-scale phase III clinical trials (FREEDOMS I and II) (FREEDOMS - FTY720 Research Evaluating Effects of Daily Oral Therapy in Multiple Sclerosis) in RRMS revealed that fingolimod, compared with placebo, reduces the ARR (48–55%) ()Citation29,Citation30. The reduction in 3-months confirmed disability progression, as measured by the EDSS vs placebo, was only significant in the FREEDOMS I study (28%)Citation29. In addition, with regard to a spectrum of MRI-specific measures, fingolimod was superior to placebo in reducing the number of new or enlarging of T2-lesions, Gad + lesions, and BVL. In a comparative study with INFβ-1a IM (TRANSFORMS - TRial Assessing injectable interferoN vS. FTY720 Oral in RRMS), fingolimod reduced the ARR at 12 months by 52%, and the number of Gad + MRI lesions by >50%Citation31. At the end of the study, 46% of fingolimod-treated patients had NEDA vs 34% with INFβ-1a. With respect to progression of disability, no significant difference was observed between the two drugsCitation31. A post-hoc analysis of the study data of the FREEDOMS trial revealed that 33% of patients treated with fingolimod showed NEDA, vs 13% of patients treated with placebo ()Citation32, while a post-hoc analysis of TRANSFORMS demonstrated that fingolimod was very effective in patients with high disease activity with an ARR reduction of 61% relative to INFβ-1aCitation33. The long-term extension studies of FREEDOMSCitation34 and TRANSFORMSCitation35 confirmed the findings from the pivotal studies. In addition, the ARR also improved in the groups that switched from placebo to fingolimodCitation34. In the fingolimod-extension groups, more patients were free from 3-month confirmed progression of disability compared with the groups of all patients who switched from placebo to fingolimod. In addition, reduced BVL was observed in the fingolimod-extension groups, while in the groups of patients who switched from placebo to fingolimod BVL improved. In the TRANSFORMS extension trial, patients re-exposed to fingolimod showed a 37% reduction in ARR vs patients who switched from INFβ-1a to fingolimodCitation35. The fact that fingolimod-extension patients from the pivotal studies had greater improvements than those who only received fingolimod in the extensions provides evidence that early initiation of fingolimod therapy is superior. Furthermore, in the TRANSFORMS extension trial, patients who were switched from INFβ-1a to fingolimod had a 50% improvement in the ARR, underlining the rationale for a switch from INFβ-1a to fingolimodCitation35. This was also confirmed in a retrospective database analysis which revealed that patients switching from INFβ to fingolimod experienced fewer relapses than those switching from INFβ to GACitation36. Similarly, in routine clinical practice, switching from DMTs to fingolimod proved to be beneficial in terms of ARR and disability progressionCitation37. With regards to the effect of fingolimod on MRI parameters, the TRANSFORMS extension also showed that fingolimod-extension patients had lower BVL and MRI activity than those patients who switched from IFN-β 1a to fingolimodCitation35. Another post-hoc analysis of study data revealed that patients who switched from INFβ-1a to fingolimod in the first year had a 50% increase in NEDACitation35.
Table 1. Summary efficacy data from pivotal placebo-controlled and active comparator phase II/III trials of disease-modifying therapies in RRMS in the FDA/EMA approved dosages.
Fingolimod induces transient symptomatic and mostly self-limiting cardiac adverse effects (bradycardia and first- and second-degree atrioventricular block). These adverse events have been observed in 0.6–1% of patients after the first-dose administrationCitation37. Therefore, first-dose administration of fingolimod requires baseline electrocardiogram, clinical observation, and monitoring for at least 6 h, until potential arrhythmia resolves (e.g. bradycardia <45 bpm). In general, fingolimod is contraindicated in patients with pre-existing cardiac disease. Similarly, fingolimod can induce a slight transient increase in blood pressure.
The presence of S1P-2 receptors in the retina may potentially lead to reversible fingolimod-induced macular edema, which occurs within the first trimester of treatmentCitation38,Citation39. This complication is more frequently observed in diabetic patients and patients with prior uveitis. In those patients, ophthalmologic examination should be done before starting therapy and follow-up controls scheduled at least annually. Lymphopenia is likely responsible for opportunistic infections, in particular, herpes virus infections and progressive multifocal leukoencephalopathy (PML), which may occur in fingolimod-treated patients. Consequently, varicella-zoster virus vaccination has been recommended prior to the initiation of therapy in patients with no history of chickenpox or immunization. As of May 2017, there have been 13 confirmed PML cases in >213,000 fingolimod-treated patients (>453,000 patient-years) who had not been previously exposed to natalizumab, and 17 cases in fingolimod-treated patients after switching from natalizumabCitation40. Other commonly observed adverse events are asymptomatic increase in liver transaminases (15–18%), cough, diarrhea, hypertension, headache, upper respiratory tract infection, and fatigueCitation30,Citation31,Citation33. Monitoring complete blood count (CBC) and hepatic function before initiating therapy and 3-monthly for 1 year, and then periodically during treatment with fingolimod are recommendedCitation37. As of February 2015, 151 cases of basal cell carcinoma in patients treated with fingolimod had been identified. Therefore, a dermatological examination at the start of the treatment, and then annually, is recommendedCitation41. Lymphocytic counts should be monitored and levels <200 cells/mm3 need discontinuation of fingolimodCitation41. Peripheral counts usually return back to normal within 1–2 months after discontinuation of the drug. Therefore, at least 2 months washout is recommended when switching from fingolimod to alemtuzumab. Switching from fingolimod to natalizumab or dimethyl fumarate requires at least 2 weeks (max = 8 weeks)Citation21.
Safety data from clinical trials and pregnancy registriesCitation42 reveal potential for fetal abnormalities, particularly when fetal exposure to the drug happened in the first trimester of pregnancy. It is, therefore, recommended that women of childbearing potential take effective contraception during fingolimod therapy and for 2 months after discontinuation.
Teriflunomide
Teriflunomide (14 mg once daily) is an active metabolite of leflunomide approved for the treatment of rheumatoid arthritisCitation43. Teriflunomide acts primarily by selective and reversible inhibition of the mitochondrial enzyme dihydro-orotate dehydrogenase (DHODH) used in activated and proliferating T and B lymphocytes for de novo pyrimidine synthesis—sparing of pyrimidine salvage pathway—and, hence, allowing maintenance of protective immunity throughout the treatmentCitation44. Ultimately it reduces the number of lymphocytes available for migration into the CNS. Since teriflunomide does not affect resting lymphocytes, the immune surveillance is not affected.
Pharmacokinetic data from healthy volunteers and MS patients show that teriflunomide is efficiently absorbed within 1–4 h (Tmax), with an oral bioavailability of almost 100%Citation45. Its absorption is unaffected by dietary intake. The drug is highly protein-bound (99%) and distributed mainly in plasma. Hydrolysis and oxidation are the major routes of biotransformation, and it is excreted, mainly unchanged, through bile. Teriflunomide is very slowly eliminated from plasma. After repeated doses, the half-life is ∼19 daysCitation43,Citation44. Elimination of the drug may be accelerated by administration of cholestyramine orally or activated charcoal orally for 11 days.
The phase III clinical trials TEMSO (TEriflunomide Multiple Sclerosis Oral) and TOWER (Teriflunomide Oral in people With relapsing-remitting MultiplE ScleRosis) in RRMS revealed that, compared with placebo, teriflunomide 14 mg oral once daily—the FDA and EMA approved doseCitation46,Citation47—reduced: the ARR with 32–36%, the rate of 12-week confirmed disability progression with 30–33%, and Gad + MRI activity ∼80% ()Citation9,Citation48. In TEMSO, teriflunomide had no significant effect on BVLCitation9. In the TENERE (TErifluNomidE and REbif) trial, oral teriflunomide 14 mg once daily demonstrated efficacy on ARR and time to first occurrence of confirmed relapse or permanent treatment discontinuation for any cause comparable with IFNβ-1a 44 µg SC three times weeklyCitation49. Treatment Satisfaction Questionnaire for Medication (TSQM) scores were also significantly higher with teriflunomide. The TOPIC (TeriflunOmide versus Placebo In patients with first Clinical symptom of multiple sclerosis) trial revealed that teriflunomide 14 mg daily—over a 108-week period—is beneficial in prolonging time to second clinical event and reduce the risk of conversion to clinical definite RRMS by 43%Citation50. In clinical isolated syndrome (CIS) with silent MRI lesions, teriflunomide significantly delayed the time to a second relapse and reduced the number of new MRI lesionsCitation50. A post-hoc analysis of TOWER study data revealed that teriflunomide 14 mg compared with placebo reduced ARR of severe relapses with 52.5%, relapses with neurological sequelae with 37%, and relapses leading to hospitalization with 34%Citation51.
The most common teriflunomide-associated adverse effects are mild elevation of alanine aminotransferase (ALT), headache, diarrhea, nausea, transient hair thinning, or decreased hair densityCitation52. Less commonly observed adverse effects are elevated blood pressure, neutropenia, and lymphopenia (transitory during the first 3–6 months of therapy) and peripheral neuropathyCitation53. In the extension of the TEMSO trial (up to 9 years), the safety profile was consistent with that of the pivotal phase III trialsCitation54,Citation55. It is recommended to monitor liver function (e.g. monthly during the first 6 months of therapy, and subsequently every 2–3 months), CBC (lymphocyte count), and blood pressure during the treatment. Importantly, there have been no reports of opportunistic infections or malignancies with teriflunomide.
Because of its long half-life, an accelerated wash-out procedure is required in the case of significant hepatotoxicity or switching from teriflunomide to another DMTCitation21.
In contrast to data from animal studies, no teratogenic signals have been detected in pregnancy registries of patients treated with teriflunomideCitation56. Despite this, teriflunomide is still labelled as pregnancy category X, and it is advised to consistently use contraception after negative pregnancy testing prior to treatment initiation in woman of childbearing potential. In thee case of unexpected pregnancy, an accelerated wash-out procedure is recommended. Similarly, in the case of impending pregnancy, washout is recommended, and a teriflunomide plasma level of <0.02 g/L following the procedure is considered safe to conceive.
Dimethyl fumarate
Dimethyl fumarate, in combination with fumaric acid esters, has been licensed in Germany as an oral drug for the treatment of psoriasis (Fumaderm)Citation57. The anti-inflammatory effects of dimethyl fumarate have been associated with reductions in circulating lymphocyte counts and impeding cell migrationCitation58, which results in a shift in response from T helper 1 (Th1) to T helper 2 (Th2). This subsequently increases the levels of anti-inflammatory cytokines (interleukin (IL)-4, IL-5, and IL-10) and causes apoptosis of activated T-cellsCitation59,Citation60. The restricted migration of activated T-cells through the blood–brain barrier is the result of down-regulation of number of adhesion molecules (intracellular adhesion molecule-1, vascular cell adhesion molecule-1, and E-selectin)Citation61,Citation62. The cytoprotective effects of dimethyl fumarate are likely mediated through the activation of the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) transcriptional pathway, which attenuates oxidative stress by up-regulating Nrf2-dependent antioxidant genesCitation62. Both the EMA and FDA have approved enteric-coated dimethyl fumarate as a first-line agent in RRMS in 2013Citation63,Citation64.
The treatment with dimethyl fumarate consists of a 120-mg starting dose BID for 7 days followed by an increase to the maintenance dose of 240 mg BID. Pharmacokinetic data derived from patients with MS and healthy volunteers reveal that, immediately following intake, dimethyl fumarate is hydrolyzed to its active metabolite, monomethyl fumarateCitation65. Monomethyl fumarate is absorbed within 2–5.5 h (Tmax), and its absorption is not affected by dietary intake. Monomethyl fumarate is bound for 27–40% to protein and is extensively metabolized. Before being taken up in the systemic circulation, dimethyl fumarate is metabolized by esterases in the gastrointestinal tract and blood. Further metabolism of monomethyl fumarate occurs independent of the CYP P450 system through the tricarboxylic acid cycleCitation65. Monomethyl fumarate, glucose, fumaric acid, and citric acid are the major metabolites found in plasma. Sixty per cent of dimethyl fumarate is excreted mainly by CO2 exhalation, and the remainder by renal and fecal elimination. The terminal half-life of monomethyl fumarate is ∼1 hCitation65.
In the phase III clinical trials DEFINE (Determination of the Efficacy and safety of oral Fumarate IN rElapsing-remitting MS) and CONFIRM (COmparator aNd an oral Fumarate In Relapsing-remitting Multiple sclerosis), oral delayed-release dimethyl fumarate reduced the ARR (44–53%), the rate of 3-month disability progression (21–38%), and Gad + MRI activity (75–94%) compared with placebo ()Citation7,Citation66. Remarkably, the effect on the EDSS progression at 2 years in CONFIRM was not significant compared with placeboCitation7. Furthermore, only the DEFINE trial demonstrated a statistically significant reduction in BVLCitation67. In the long-term extension of DEFINE/CONFIRM (ENDORSE), a minimum of 5 years of treatment with the drug was associated with continued benefit (63%, 73%, and 88% of patients being free of new or enlarging T2 hyperintense lesions, new T1 hypointense lesions, and Gad + lesions, respectively) with no new/worsening in its safety and tolerability profileCitation68 A post-hoc analysis from the DEFINE and CONFIRM study data revealed that, after 2 years of treatment a higher proportion of patients (23%) on dimethyl fumarate achieved NEDA compared with only 12% in the placebo groupCitation69,Citation70.
The most commonly reported adverse events are transient gastrointestinal symptoms (nausea, upper abdominal pain, and diarrhea) and hot flushes, which typically appear 30 min after intake of the drug and subside within 90 minCitation7,Citation66. In addition, elevations of liver enzymes and lymphopenia have been reportedCitation7,Citation66,Citation71. Dimethyl fumarate-induced lymphopenia (<500 cells/mm3)—particularly if present over an extended period of time—has been considered as a potential risk factor for PMLCitation72. Five cases of PML have been reported while on dimethyl fumarateCitation72. The FDA and EMA recommend to monitor LFTs and CBC (in particular lymphocyte count) every 3 months during the treatment, and to consider discontinuation of dimethyl fumarate in patients with lymphocyte counts <500 cells/mm3 persisting for more than 6 months, particularly in anti-John Cunningham (JC) virus antibody positive patientsCitation72,Citation73.
In view of the short half-life of dimethyl fumarate, no washout period is required when patients switch from dimethyl fumarate to alemtuzumab, natalizumab, or fingolimod, provided the lymphocyte count is normalCitation21.
Pregnancy registries of patients treated with dimethyl fumarate have not revealed any increased incidence of spontaneous abortion or teratogenic signalsCitation74. Dimethyl fumarate is labeled as pregnancy category C.
Cladribine
In August 2017, the EMA approved cladribine as an oral DMT for the treatment of patients with RRMS with highly active disease, which includes patients in need of treatment escalation after breakthrough disease while on IFNβ or GACitation75,Citation76.
Cladribine, an adenosine deaminase-resistant, purine nucleoside analog of deoxyadenosine is phosphorylated intracellularly by deoxycytidine kinase to its active form, deoxynucleotide monophosphateCitation77,Citation78. In cells with a high ratio of deoxycytidine kinase to deoxynucleotidase, e.g. lymphocytes and monocytes, the metabolite accumulates and is converted to active triphosphate deoxynucleotide, which interferes with cellular metabolism and ultimately causes apoptosis. Clinically translated, cladribine induces a rapid and prolonged decrease in circulating B, CD4+, and CD8+ T-cells, which is sustained for at least 6–12 monthsCitation77,Citation79.
In MS patients, cladribine is rapidly (Tmax = 30–50 min) but incompletely absorbed after oral intake (bioavailability of 37–51%)Citation79. Dietary intake has little influence on its absorption. The drug has a low protein binding (0–20%)Citation79, and its half-life varies between 5.6–19.7 h, referring to different phases of elimination. Cladribine has the ability to cross the blood–brain barrier, which provides the potential to reduce lymphocytes at the site of the focal inflammation in the CNSCitation79. Studies also revealed that cladribine impedes the influx of autoreactive T-cells into the CNSCitation80. The main metabolite in plasma, 2-chloroadenine, which is also cytotoxic, likely contributes to the clinical effect of the drugCitation81. Around 25% of the drug is excreted unchanged in the urine. Renal elimination of cladribine correlates with the creatinine clearanceCitation82. The EMA approved and recommended the cumulative dose regimen of cladribine, of 3.5 mg/kg over 2 years. This is administered as one treatment course of 1.75 mg/kg per year (e.g. a 70 kg person will receive 60 mg week 1 and 60 mg week 2 (with 1 month interval) the first year and again the same dose regimen the second year)Citation76. Each treatment course consists of two treatment weeks, one at the beginning of the first month and one at the beginning of the second month of the respective treatment year. Each treatment week consists of 4 or 5 days on which a patient receives 10 mg or 20 mg (one or two tablets) as a single daily dose, depending on the patient’s body weight. Following completion of the two treatment courses, no further cladribine treatment is required in years 3 and 4. Re-initiation of therapy after year 4 has not been studied.
Cladribine was granted EMA approval for RRMS based on the results of three clinical trials comparing oral cladribine vs placebo (CLARITY)Citation83, oral cladribine plus IFNβ vs placebo plus IFNβ (ONWARD)Citation84, and oral cladribine vs placebo (CLARITY Extension)Citation85. In the CLARITY (CLAdRIbine Tablets treating MS orallY) trial, oral cladribine—in the EMA approved dosage (3.5 mg/kg)—reduced the ARR (58%), the rate of 3-month sustained disability progression (33%), active T2-weighted lesions (73%), and Gad + MRI activity (86%) compared with placeboCitation83. Notably, approximately one third of patients participating in the CLARITY trial had previously been treated with another DMT (). The proportion of patients remaining relapse-free at 96 weeks (77.8%) was significantly higher (19%) compared with placebo. A post-hoc analysis from the CLARITY study data revealed that, after 96 weeks of treatment, 44% of patients on cladribine achieved NEDA compared with 16% in the placebo groupCitation86, an effect which was observed in all patient sub-groups, including those with high disease activity. Cladribine also significantly reduced BVL (19%)Citation87.
The ONWARD (Oral cladribine added oN to interferon beta (IFNβ) in patients With Active Relapsing Disease) study—presented as abstract only—revealed a 63% reduction in ARR in the cladribine (3.5 mg/kg)-IFNβ group compared to the placebo-IFNβ armCitation84, and the corresponding mean number of T1 Gad + lesions per scan was 82% lower ().
Almost 74% of patients from the original CLARITY trial went—with variable gap periods in between—into the CLARITY extension trialCitation85. This 2-year extension phase showed a 33% reduction in ARR in those patients who received cladribine (3.5 mg/kg) during both the original study (CLARITY) and the CLARITY extension phase compared with those treated with cladribine in the original study and placebo in the CLARITY extension phaseCitation85. The proportion of patients qualifying relapse-free was 75.6% in the CLARITY extension study, which was similar to that in the CLARITY trial (77.8%). The proportion of patients without Gad + lesions was 85–90% in cladribine-treated groups in the CLARITY extension study vs 73–80% in placebo-treated groups, while the respective proportions without active T2 lesions were 38–44% and 28–34%. The mean numbers of new Gad + lesions/subject/scan were 89% lower in patients treated with cladribine in both CLARITY and CLARITY extension trials compared with those treated with cladribine in CLARITY and placebo in the CLARITY extension studyCitation88. The CLARITY extension study highlighted the durability of the clinical response. Despite the lymphocyte recovery there was no evidence of increased disease activity after the treatment was completed. This supports the rationale for early treatment with the drug in the absence of significant side-effects. Lastly, there appeared to be no incremental benefit from additional treatment with cladribine in years 3 and 4, following the initial courses in years 1 and 2Citation85.
The phase III ORACLE MS (ORAl CLadribine in Early MS) trial in CIS was prematurely terminated by Merck on safety concerns expressed by the Regulatory AuthoritiesCitation89. The analysis of the data from the patients enrolled revealed that cladribine (3.5 mg/kg) significantly delayed the time to conversion to clinical definite MS, by risk reduction of 67% relative to placebo. Cladribine also significantly reduced risk of conversion to ‘McDonald MS’ vs placebo with 50%. There was a 89% risk reduction for developing new or persisting T1 Gad + lesions and 79% for new or enlarging T2 lesions compared with placebo.
Owing to its mechanism of action, the most commonly reported adverse event is lymphopenia (T-cells, B-cells, and natural killer cells) (12–22%), which reached a nadir at 3–4 weeks following the first treatment courseCitation90. In the CLARITY study, the median lymphocyte counts at week 96 were 56% of baseline valuesCitation83. Grade 3 and 4 lymphopenia (<500 cells/mm3) was reported in 2–26% of patients treated with cladribine 3.5 mg/kg (vs 0.5% in the placebo group). However, in the ONWARD study the frequency of lymphopenia was high in the double-blind phase of the trial (grade 3 toxicity was observed in 62% in the cladribine 3.5 mg/kg-IFNβ arm, while only 2% in the placebo-IFNβ arm), which was much higher than previously recorded for cladribine therapy alone (25%)Citation91. Grade 4 lymphopenia rates were low (<5%) in the double-blind phase. No cases of PML, and no unexpected disseminated or invasive infections have been reported in patients treated for RRMS. Malignancy was observed in 0.34% of patients when treated with cladribineCitation90.
Animal studies revealed evidence of teratogenicity, embryotoxicity, and fetotoxicity at doses equivalent to those recommended in humansCitation92. Cladribine is labeled as pregnancy category D. As cladribine interferes with DNA synthesis, adverse effects on gametogenesis should be strongly considered. Therefore, prior to treatment initiation—both in year 1 and year 2—women of childbearing potential and males who wish to father a child should be counseled regarding the potential for teratogenic effects and the need for effective contraceptionCitation76. Prior to treatment initiation with cladribine—both in year 1 and year 2—pregnancy must be excluded, inclusing preventive measures with effective contraception during treatment and ≥6 months after the last doseCitation76. Pregnancy during the therapy should prompt discontinuation of the treatment. Similarly, breastfeeding should not be undertaken during treatment and for 6 months after the last dose.
When considering switching to or from cladribine treatment, there will be a potential additive effect on the immune system. Switching should be considered safe when lymphocyte counts have returned to normal. When switching from other DMTs, a baseline MRI is advisedCitation76.
Therapeutic strategies and guidelines
There is evidence that relapses occurring within the first 2 years of the onset of clinical disease are the principal determinant of early disease progression and long-term disability, justifying a “low threshold” strategy in its managementCitation93. Factors influencing the therapeutic decision-making process in RRMS are: recent disease activity (relapse rate, severity, and recovery), the degree of neurological impairment, the MRI lesion load, and the presence of Gad + brain and spinal cord lesions, anti-JC virus antibody status, drug availability and cost, adverse effect profiles, tolerability, comorbid illnesses and medications, monitoring requirements, attitudes, lifestyle factors, and, last but not least, patient preferences (reproductive considerations; desire to avoid self-injections; or specific adverse effects)Citation21,Citation94. Consequently, a customized and personalized approach based on benefit-risk assessment, tolerability, convenience, and patient preference is essential for the successful management of RRMS, and must be consistent with the clinical stage and course of the disease.
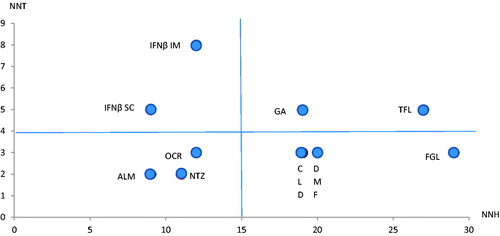
The main findings from the pivotal clinical trials of the oral DMTs are summarized in . Unfortunately, no head-to-head trials have been performed amongst the oral DMTs. shows the effectiveness and safety of the oral DMTs vs non-oral DMTs, expressed by the number needed to treat vs the number needed to harmCitation21,Citation95. The graph shows that three of the oral DMTs are in the right lower quadrant (low NNT, and high NNH) and one (teriflunomide) is in the right upper quadrant (slightly higher NNT, but still high NNH). This confirms their efficacy and safety compared to non-oral DMTs particularly in active RRMS.
Figure 1. Plot of the number needed to treat (NNT) vs number needed to harm (NNH) for all currently approved disease-modifying therapies (DMTs). The figure shows that the oral DMTs belong, except for teriflunomide, to the right lower quadrant, which reflects a low number needed to treat (NNT) and a high number needed to harm (NNH) (indicating relative good efficacy with good safety profile) relative to the parenteral DMTs. The data were extracted from Deleu et al.Citation21 and Siddiqui et al.Citation95. Abbreviations. ALM, Alemtuzumab; CLD, Cladribine; DMF, Dimethyl fumarate; FGL, Fingolimod; GA, Glatiramer acetate; IFNβ IM, interferon beta intramuscular; IFNβ SC, interferon beta subcutaneous; NTZ, Natalizumab; OCR, Ocrelizumab; TFL, teriflunomide.
Taking together the provided efficacy, safety, and tolerability data, and the corresponding approval by the FDA and EMA, the following recommendations in adults with CIS or RRMS can be provided:
Teriflunomide has a convenient once daily dosing. Unfortunately, teriflunomide has not yet received approval in CIS by FDA or EMA.
Based on similar efficacy as injectable DMTs, teriflunomide is considered a first-line oral DMT for active RRMS. As appropriate wash-out procedures are available, the drug can be eliminated quickly in the case of adverse effects, prior to pregnancy or switch to another DMT.
Dimethyl fumarate is considered a first-line DMT for active RRMS, but with higher efficacy than teriflunomide. Since PML has been reported, dimethyl fumarate therapy requires monitoring of lymphocytic count.
Fingolimod is approved by FDA as first-line DMT in active RRMS and both by FDA and EMA for patients with rapidly evolving severe or highly active RRMS with sub-optimal response to at least one DMT.
Fingolimod is the first choice in anti-JC virus antibody negative patients with highly active or rapidly evolving RRMS.
Cladribine is very effective in patients with RRMS with highly active disease and may be considered as an induction therapy due to its sustained immunological effects after clearance of the drug.
The prolonged lymphopenia following cladribine treatment has so far not resulted in an increased incidence of opportunistic infections or malignancies.
Availability of oral DMTs and current guidelines in the MENA region
The oral DMTs have been launched in several countries of the MENA region (), with fingolimod being the most widely available one. Cladribine has not been registered yet in any of the MENA countries. As for the majority of the Arabian Gulf states (Kuwait, Saudi Arabia, Bahrain, Qatar, Oman, and United Arab Emirates) once the drug has been approved by the FDA or EMA, the drug can be prescribed and imported on a patient named-basis.
Table 2. The oral disease-modifying drugs launched in several countries of the MENA region.
Guidelines on the use of DMTs for MS have been published from the MENACTRIMS and a few countries in the MENA region (Kuwait and Qatar)Citation21,Citation96,Citation97.
Both in the MENACTRIMS guidelines published in 2015Citation96 and the consensus recommendation from Kuwait published in 2016Citation97, teriflunomide and dimethyl fumarate were positioned—together with the interferons and GA—as first-line agents in treatment-naïve patients with “non-aggressive” RRMS. The latter defined as having one disabling relapse in the last year, or an active MRI containing new or Gad + lesions that have developed during the last year. Furthermore, fingolimod was recommended when treatment escalation is required because of needle phobia, adverse drug reactions, contraindications, or sub-optimal response to these first-line DMTs (interferons, GA, teriflunomide, or dimethyl fumarate). In aggressive RRMS—defined as MS with rapidly evolving severe relapsing remitting course evidenced by ≥2 disabling relapses in the previous year and ≥1 Gad + lesion or significant increase in T2 lesion load on MRI—fingolimod is aligned together with the intravenous DMTs, but ultimately the choice of DMT is based on the risk stratification (serum JC virus antibody status, prior immunosuppressant use, cardiac disease, diabetes, retinal disorders, previous autoimmune diseases, and thyroid disorders). Cladribine was not included yet in the MENACTRIMS and Kuwait guidelines. According to the recommendations in Qatar published in 2017, teriflunomide, dimethyl fumarate, and, if required, fingolimod are recommended as first-line DMTs in active RRMS in treatment-naïve patients and patients with needle phobia, adverse drug reactions (ADRs), contraindications, or sub-optimal response to IFNβ or GACitation21. In patients with rapidly evolving or highly active RRMS—whether treatment-naïve or not—and based on risk stratification (serum JCV antibody, prior immunosuppressant use, cardiac disease, diabetes, retinal disorders, previous autoimmune diseases, and thyroid disorders), fingolimod or intravenous DMTs were recommended. In the case of sub-optimal treatment response of the fingolimod or intravenous DMTs, cladribine was recommended in the guideline—which was published prior to EMA approval—as a treatment escalationCitation21.
Conclusion
The current available injectable DMTs for RRMS are affected by sub-optimal patient adherence. The approval by the FDA and EMA of several oral DMTs for RRMS—with a different mechanism of action—has significantly reduced the burden of drug administration, has promoted patient satisfaction, and has improved patient adherence. Post-hoc analyses of study data from individual pivotal clinical trials of fingolimod, dimethyl fumarate, teriflunomide and cladribine revealed comparable effects for these oral DMTs across key clinical outcomes (ARR, disability progression, and MRI activity), as determined by NNT evaluations. The ever changing therapeutic landscape of RRMS—with more aggressive and earlier treatment in the disease course—comes, however, with new challenges. Safety profiles vary dependent on their mechanism of action, and more attention goes in the monitoring of autoimmune disease and potential development of tumors.
With the exception of fingolimod, the limited distribution, and high cost of these oral DMTs in the MENA region remains a major challenge.
Transparency
Declaration of funding
This manuscript was not funded.
Declaration of financial/other interests
DD received honoraria for presentations at meetings and served in advisory boards of Novartis, Sanofi, Biologix, and Merck. All other authors have disclosed that they have no significant relationships with or financial interests in any commercial companies related to this study or article. CMRO peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
None reported.
References
- Bergvall N, Petrilla AA, Karkare SU, et al. Persistence with and adherence to fingolimod compared with other disease-modifying therapies for the treatment of multiple sclerosis: a retrospective US claims database analysis. J Med Econ 2014;17:696-707
- Halpern R, Agarwal S, Dembek C, et al. Comparison of adherence and persistence among multiple sclerosis patients treated with disease-modifying therapies: a retrospective administrative claims analysis. Patient Prefer Adherence 2011;5:73-84
- Agashivala N, Wu N, Abouzaid S, et al. Compliance to fingolimod and other disease modifying treatments in multiple sclerosis patients, a retrospective cohort study. BMC Neurol 2013;13:138
- Giovannoni G, Southam E, Waubant E. Systematic review of disease modifying therapies to assess unmet needs in multiple sclerosis: tolerability and adherence. Mult Scler 2012;18:932-46
- Hupperts R, Ghazi-Visser L, Martins Silva A, et al. The STAR study: a real-world, international, observational study of the safety and tolerability of, and adherence to, serum-free subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis. Clin Ther 2014;36:1946-57
- Treadaway K, Cutter G, Salter A, et al. Factors that influence adherence with disease-modifying therapy in MS. J Neurol 2009;256:568-76
- Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med 2012;367:1087-97
- Kappos L, Antel J, Comi G, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med 2006;355:1124-40
- O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med 2011;365:1293-303
- Gilenya. fingolimod. East Hanover: Novartis Pharmaceuticals Corporation; 2014
- Aubagio. teriflunomide. Cambridge: Genzyme Corporation; 2014
- Tecfidera. dimethyl fumarate. Cambridge: Biogen Idec Inc; 2015
- Heydarpour P, Khoshkish S, Abtahi S, et al. Multiple sclerosis epidemiology in Middle East and North Africa: a systematic review and meta-analysis. Neuroepidemiology 2015;44:232-44
- Alroughani R, Ahmed SF, Behbahani R, et al. Increasing prevalence and incidence rates of multiple sclerosis in Kuwait. Mult Scler 2014;20:543-7
- Deleu D, Mir D, Al Tabouki A, et al. Prevalence, demographics and clinical characteristics of multiple sclerosis in Qatar. Mult Scler 2013;19:816-19
- Inshasi J, Thakre M. Prevalence of multiple sclerosis in Dubai, United Arab Emirates. Int J Neurosci 2011;121:393-8
- Alshubaili AF, Alramzy K, Ayyad YM, et al. Epidemiology of multiple sclerosis in Kuwait: new trends in incidence and prevalence. Eur Neurol 2005;53:125-31
- Giovannoni G, Turner B, Gnanapavan S, et al. Is it time to target no evident disease activity (NEDA) in multiple sclerosis? Mult Scler Relat Disord 2015;4:329-33
- Havrdova E, Galetta S, Hutchinson M, et al. Effect of natalizumab on clinical and radiological disease activity in multiple sclerosis: a retrospective analysis of the natalizumab safety and efficacy in relapsing-remitting multiple sclerosis (AFFIRM) study. Lancet Neurol 2009;8:254-60
- Kappos L, De Stefano N, Freedman MS, et al. Inclusion of brain volume loss in a revised measure of ‘no evidence of disease activity’ (NEDA-4) in relapsing-remitting multiple sclerosis. Mult Scler 2016:22;1297-305
- Deleu D, Mesraoua B, El Khider H, et al. Optimization and stratification of multiple sclerosis treatment in fast developing economic countries: a perspective from Qatar. Curr Med Res Opin 2017;33:439-58
- Dubey D, Cano CA, Stuve O. Intractable and highly active relapsing multiple sclerosis - role of alemtuzumab. Neuropsychiatr Dis Treat 2015;11:2405-14
- EMA Gilenya (fingolimod). Summary of product characteristics (fingolimod). London, UK: EMA; 2011. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002202/WC500104528.pdf [Last accessed 30 November 2017]
- US FDA. GILENYA (fingolimod); highlights of prescribing information. Maryland, USA: FDA; 2012. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/022527s008lbl.pdf [Last accessed 14 July 2017]
- Brinkmann V. FTY720 (fingolimod) in multiple scerosis: therapeutic effects in the immune and the central nervous system. Br J Pharmacol 2009;158:1173-82
- Miron VE, Jung CG, Kim HJ, et al. FTY720 modulates human oligodendrocyte progenitor process extension and survival. Ann Neurol 2008;63:61-71
- Ingwersen J, Aktas O, Kuery P, et al. Fingolimod in multiple sclerosis: mechanisms of action and clinical efficacy. Clin Immunol 2012;142:15-24
- David OJ, Kovarik JM, Schmouder RL. Clinical pharmacokinetics of fingolimod. Clin Pharmacokinet 2012;51:15-28
- Kappos L, Radue EW, O’Connor P, et al. A placebo controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010;362:387-401
- Calabresi PA, Radue EW, Goodin D, et al. Safety and efficacy of fingolimod in patients with relapsing remitting multiple sclerosis (FREEDOMS II): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol 2014;13:545-56
- Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med 2010;362:40215
- Kappos L, Radue EW, O’Connor P, et al. Fingolimod treatment increases the proportion of patients who are free from disease activity in multiple sclerosis: results from a phase 3, placebo-controlled study (FREEDOMS). Neurology 2011;76(Suppl 4):A563
- Cohen JA, Barkhof F, Comi G, et al. Fingolimod versus intramuscular interferon in patient subgroups from TRANSFORMS. J Neurol 2013;260:2023-32
- Kappos L, O’Connor P, Radue EW, et al. Long-term effects of fingolimod in multiple sclerosis: the randomized FREEDOMS extension trial. Neurology 2015;84:1582-91
- Cohen JA, Khatri B, Barkhof F, et al. Long-term (up to 4.5 years) treatment with fingolimod in multiple sclerosis: results from the extension of the randomised TRANSFORMS study. J Neurol Neurosurg Psychiatry 2016;87:468-75
- Bergvall N, Makin C, Lahoz R, et al. Relapse rates in patients with multiple sclerosis switching from interferon to fingolimod or glatiramer acetate: a US claims database study. PLoS One 2014;9:e88472
- Ziemssen T, Schwarz HJ, Fuchs A, et al. 36 month PANGAEA: a 5-year non-interventional study of safety, efficacy and pharmacoeconomic data for fingolimod patients in daily clinical practice. Neurology 2015;84(Suppl):P3.251
- Novartis Pharma GmbH. Gilenya SmPC. London, UK: EMA; 2011. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002202/WC500104528.pdf [Last accessed 29 November 2017]
- Zarbin MA, Jampol LM, Jager RD, et al. Ophthalmic evaluations in clinical studies of fingolimod (FTY720) in multiple sclerosis. Ophthalmology 2013;120:1432-9
- Guarnera C, Bramanti P, Mazzon E. Comparison of efficacy and safety of oral agents for the treatment of relapsing-remitting multiple sclerosis. Drug Des Devel Ther 2017;11:2193-207
- EMA. New recommendations to minimise risks of the rare brain infection PML and a type of skin cancer with Gilenya. London, UK: EMA; 2015. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2015/12/news_detail_002447.jsp&mid=WC0b01ac058004d5c1 [Last accessed 12 December 2017]
- Karlsson G, Francis G, Koren G, et al. Pregnancy outcomes in the clinical development program of fingolimod in multiple sclerosis. Neurology 2014;82:674-80
- Osiri M, Shea B, Robinson V, et al. Leflunomide for treating rheumatoid arthritis. Cochrane Database Syst Rev 2003;(1):CD002047
- Bar-Or A, Pachner A, Menguy-Vacheron F, et al. Teriflunomide and its mechanism of action in multiple sclerosis. Drugs 2014;74:659-74
- Wiese MD, Rowland A, Polasek TM, et al. Pharmacokinetic evaluation of teriflunomide for the treatment of multiple sclerosis. Expert Opin Drug Metab Toxicol 2013;9:1025-35
- European Medicines Agency (EMA). Teriflunomide Summary of product characteristics. London, UK: EMA; 2017. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002514/WC500148682.pdf [Last accessed 30 November 2017]
- US FDA. AUBAGIO (teriflunomide); highlights of prescribing information. Maryland, USA: FDA; 2012. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202992s000lbl.pdf [Last accessed 30 November 2017]
- Confavreux C, O’Connor P, Comi G, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol 2014;13:247-56
- Vermersch P, Czlonkowska A, Grimaldi LM, et al. Teriflunomide versus subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis: a randomised, controlled phase 3 trial. Mult Scler 2014;20:705-16
- Miller AE, Wolinsky JS, Kappos L, et al. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): a randomised, double blind, placebo-controlled, phase 3 trial. Lancet Neurol 2014;13:977-86
- Miller AE, Macdonell R, Comi G, et al. Teriflunomide reduces relapses with sequelae and relapses leading to hospitalizations: results from the TOWER study. J Neurol 2014;261:1781-8
- Comi G, Freedman MS, Kappos L, et al. Pooled safety and tolerability data from four placebo-controlled teriflunomide studies and extensions. Mult Scler Relat Disord 2016;5:97-104
- Chan A, de Seze J, Comabella M. Teriflunomide in patients with relapsing-remitting forms of multiple sclerosis. CNS Drugs 2016;30:41-51
- Papadopoulou A, Kappos L, Sprenger T. Safety of teriflunomide for the management of relapsing-remitting multiple sclerosis. Expert Opin Drug Saf 2015;14:749-59
- O’Connor P, Comi G, Freedman MS, et al. Long-term safety and efficacy of teriflunomide: nine-year follow-up of the randomized TEMSO study. Neurology 2016;86:920-30
- Kieseier BC, Benamor M. Pregnancy outcomes following maternal and paternal exposure to teriflunomide during treatment for relapsing-remitting multiple sclerosis. Neurol Ther 2014;3:133-8
- Moharregh-Khiabani D, Linker RA, Gold R, et al. Fumaric acid and its esters: an emerging treatment for multiple sclerosis. Curr Neuropharmacol 2009;7:60-4
- Schilling S, Goelz S, Linker R, et al. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clin Exp Immunol 2006;145:101-7
- Treumer F, Zhu K, Gläser R, et al. Dimethylfumarate is a potent inducer of apoptosis in human T cells. J Invest Dermatol 2003;121:1383-8
- Cannella B, Cross AH, Raine CS. Adhesion-related molecules in the central nervous system. Upregulation correlates with inflammatory cell influx during relapsing experimental autoimmune encephalomyelitis. Lab Invest 1991;65:23-31
- Vandermeeren M, Janssens S, Borgers M, et al. Dimethylfumarate is an inhibitor of cytokine-induced E-selectin, VCAM-1, and ICAM-1 expression in human endothelial cells. Biochem Biophys Res Commun 1997;234:19-23
- di Nuzzo L, Orlando R, Nasca C, et al. Molecular pharmacodynamics of new oral drugs used in the treatment of multiple sclerosis. Drug Des Devel Ther 2014;8:555-68
- European Medicines Agency (EMA). Dimethyl fumarate; summary of product characteristics. London, UK: EMA; 2014. Available at: http://www.ema.europa.eu/docs/it_IT/document_library/EPAR_-_Product_Information/human/002601/WC500162069.pdf [Last accessed 30 November 2017]
- US FDA. TECFIDERA™ (dimethyl fumarate); highlights of prescribing information. Maryland, USA: FDA; 2013. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/204063lbl.pdf [Last accessed 30 November 2017]
- Burness CB, Deeks ED. Dimethyl fumarate: a review of its use in patients with relapsing-remitting multiple sclerosis. CNS Drugs 2014;28:373-87
- Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med 2012;367:1098-107
- Arnold DL, Gold R, Kappos L, et al. Effects of delayed-release dimethyl fumarate on MRI measures in the Phase 3 DEFINE study. J Neurol 2014;261:1794-802
- Gold R, Arnold DL, Bar-Or A, et al. Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: interim analysis of ENDORSE, a randomized extension study. Mult Scler 2017;23:253-65
- Havrdova E, Gold R, Fox R, et al. BG-12 (dimethyl fumarate) treatment for relapsing-remitting multiple sclerosis (RRMS) increases the proportion of patients free of measured clinical and neuroradiologic disease activity in the phase 3 studies. Neurology 2013;80(Suppl):P07.106
- Havrdova E, Giovannoni G, Gold R, et al. Effect of delayed-release dimethyl fumarate on no evidence of disease activity in relapsing-remitting multiple sclerosis: integrated analysis of the phase III DEFINE and CONFIRM studies. Eur J Neurol 2017;24:726-33
- Longbrake EE, Naismith RT, Parks BJ, et al. Dimethyl fumarate-associated lymphopenia: Risk factors and clinical significance. Mult Scler J Exp Transl Clin 2015;1:1-8
- FDA Drug Safety Communication. FDA warns about case of rare brain infection PML with MS drug Tecfidera (dimethyl fumarate). Maryland, USA: FDA; 2014. Available at: https://www.fda.gov/Drugs/DrugSafety/ucm424625.htm. [Last accessed 30 November 2017]
- EMA recommendation: Updated recommendations to minimise the risk of the rare brain infection PML with Tecfidera. London, UK: EMA; 2015. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2015/10/news_detail_002423.jsp&mid=WC0b01ac058004d5c1 [Last accessed 30 November 2017]
- Gold R, Phillips JT, Havrdova E, et al. Delayed-release dimethyl fumarate and pregnancy: preclinical studies and pregnancy outcomes from clinical trials and postmarketing experience. Neurol Ther 2015;4:93-104
- European Medicines Agency [Internet]. EPAR summary for the public. Mavenclad. London, UK: EMA; 2017. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/004230/WC500234564.pdf [Last accessed 30 November 2017]
- European Medicines Agency [Internet]. Mavenclad Summary of Product Characteristics. London, UK: Merck Serono Europe Limited; 2017. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004230/WC500234561.pdf [Last accessed 30 November 2017]
- Beutler E. Cladribine (2-chlorodeoxyadenosine). Lancet 1992;340:952-6
- Hartung HP, Aktas O, Kieseier B, et al. Development of oral cladribine for the treatment of multiple sclerosis. J Neurol 2010;257:163-70
- Liliemark J. The clinical pharmacokinetics of cladribine. Clin Pharmacokinet 1997;32:120-31
- Kopadze T, Dobert M, Leussink VI, et al. Cladribine impedes in vitro migration of mononuclear cells: a possible implication for treating multiple sclerosis. Eur J Neurol 2009;16:409-12
- Lindemalm S, Liliemark J, Juliusson G, et al. Cytotoxicity and pharmacokinetics of cladribine metabolite, 2-chloroadenine in patients with leukemia. Cancer Lett 2004;210:171-7
- Savic RM, Novakovic AM, Ekblom M, et al. Population pharmacokinetics of cladribine in patients with multiple sclerosis. Clin Pharmacokinet 2017;56:1245-53
- Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. New Engl J Med 2010;362:416-26
- National Institutes of Health, Clinicaltrials.com. A phase 2 study of cladribine add-on to interferon-beta (IFN-beta) therapy in multiple sclerosis (MS) subjects with active disease (ONWARD). Bethesda, USA: NLM; 2007. Available at: https://clinicaltrials.gov/ct2/show/NCT00436826?term=cladribine+and+multiple+sclerosis&rank = 3 [Last accessed 30 November 2017]
- Giovannoni G, Soelberg Sorensen P, Cook S, et al. Safety and efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis: results from the randomized extension trial of the CLARITY study. Mult Scler 2017 Aug 1:1352458517727603. doi: 10.1177/1352458517727603
- Giovannoni G, Cook S, Rammohan K, et al. Sustained disease-activity free status in patients with relapsing-remitting multiple sclerosis treated with cladribine tablets in the CLARITY study: a post-hoc and subgroup analysis. Lancet Neurol 2011;10:329-37
- De Stefano N, Giorgio A, Battaglini M, et al. Reduced brain atrophy rates are associated with lower risk of disability progression in patients with relapsing multiple sclerosis treated with cladribine tablets. Mult Scler 2018;24(2):222–226
- Comi G, Giovannoni G, Cook S, et al. Magnetic resonance imaging (MRI) outcomes in patients with relapsing-remitting multiple sclerosis (RRMS) treated with cladribine tablets: results from the 120-week phase IIIb extension of the CLARITY study. Neurology 2016;86(16 Suppl):P2.11
- Leist TP, Comi G, Cree BA, et al. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurol 2014;13:257-67
- Cook S, Vermersch P, Comi G, et al. Safety and tolerability of cladribine tablets in multiple sclerosis: the CLARITY (CLAdRIbine Tablets treating multiple sclerosis orallY) study. Mult Scler 2011;17:578-93
- Montalban X, Cohen B, Leist T, et al. Safety and tolerability of cladribine tablets added to IFN-beta therapy in patients with active relapsing multiple sclerosis: final results from the ONWARD study (amended protocol). Eur J Neurol 2016;23(Suppl S2):345-600, P21132
- European Medicines Agency, Committee for Medical Products for Human Use. Withdrawal assessment report for Movectro. EMA; 2011. London, UK: EMA; 2011. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Application_withdrawal_assessment_report/2011/03/WC500104393.pdf [Last accessed 30 November 2017]
- Scalfari A, Neuhaus A, Degenhardt A, et al. The natural history of multiple sclerosis, a geographically based study 10: relapses and long-term disability. Brain 2010;133:1914-29
- Gajofatto A, Bacchetti P, Grimes B, et al. Switching first-line disease-modifying therapy after failure: impact on the course of relapsing–remitting multiple sclerosis. Mult Scler 2009;15:50-8
- Siddiqui MK, Khurana IS, Budhia S, et al. Systematic literature review and network meta-analysis of cladribine tablets versus alternative disease-modifying treatment for relapsing-remitting multiple sclerosis. Curr Med Res Opin 2017:1-11 (doi: 10.1080/03007995.2017.1407303)
- Yamout B, Alroughani R, Al-Jumah M, et al. Consensus recommendations for the diagnosis and treatment of multiple sclerosis: the Middle East North Africa Committee for Treatment and Research In Multiple Sclerosis (MENACTRIMS). Curr Med Res Opin 2015;31:1349-61
- Alroughani R, Ashkanani A, Al-Hashel J, et al. Consensus recommendations for the diagnosis and treatment of multiple sclerosis in Kuwait. Clin Neurol Neurosurg 2016;143:51-64