
Abstract
Introduction
Phenylketonuria (PKU) is a rare autosomal recessive disorder caused by a deficiency of phenylalanine hydroxylase (PAH). Its prevalence is estimated to be 1:10,000 in Europe. PKU is the commonest congenital inborn error of metabolism. The aim of our study was to investigate the characteristics of clinical practice in relation to PKU in Italy, in order to raise awareness about the current management and therapeutic approaches adopted.
Methods
Six Italian experts conducted a systematic literature review as well as an internal survey to investigate the relevant clinical aspects. Collectively, the expert panel managed a total of 678 PKU patients treated in the early stages of the condition over a 16-year period across six centers.
Results
The management of PKU varied markedly between centers, with differences in the composition of the multidisciplinary team, dietary treatments, compliance and adherence to management, tetrahydrobiopterin use, and patient follow-up. Patients were mostly managed by a pediatric reference center from the initial PKU diagnosis during newborn screening until adulthood, without transition to a specialized adult clinician. Fogginess, concentration reduction, low attention, anxiety, irritability, memory deficit, headache, and unstable mood were common features in patients with uncontrolled blood phenylalanine levels (generally above 600 µmol/L).
Conclusion
A homogeneous and shared approach to the management of PKU patients is important. Our survey demonstrates the current management of PKU in Italy, with the aim of promoting the implementation of therapeutic strategies and follow-up, increased patient compliance and adherence, and the achievement of the phenylalanine level targets recommended by European Union guidelines. Emerging therapies are likely to become a standard treatment for patients unable to comply with diet therapy and maintain their phenylalanine levels below the threshold values.
Supplemental data for this article is available online at https://doi.org/10.1080/03007995.2020.1847717.
Introduction
Phenylketonuria (PKU) is a rare autosomal recessive disorder caused by a deficiency of phenylalanine hydroxylase (PAH). Its prevalence varies widely among countries, but is ∼1:10,000 in Europe, with a higher rate in some countries including ItalyCitation1.
PAH catalyzes the conversion of the essential amino acid phenylalanine (Phe) into tyrosine (Tyr)Citation2. Depending on the type of genetic mutation, the enzyme may be not translated at all or may have several degrees of impaired function. As a result, phenylalanine accumulates in the blood leading to a variety of clinical manifestations, including brain damage, autism, seizures, and developmental problems. In patients with PKU, every 4-week delay in starting treatment results in a decline of approximately four intelligence quotient (IQ) points, reinforcing the fact that neurological damage in PKU starts in the first few days after birth. Therefore, the organization of newborn screening and the referral to PKU centers is strongly recommended so that treatment is started no later than 10 days of ageCitation3.
A simple screening test was developed in the 1960sCitation3 and became a mandatory part of newborn screening tests in Italy in 1992 (https://www.osservatorioscreening.it/fenilchetonuria-screening-neonatale/). Elevated blood Phe levels are common in other conditions, including a high natural protein intake, prematurity, tetrahydrobiopterin (BH4) defects, and liver diseases, all of which should be ruled out prior to confirming the diagnosis of PKUCitation3. Early detection and treatment are of the utmost importance, helping to prevent intellectual disability and facilitating a normal or near-normal quality of life and IQCitation3.
Genotyping has revealed more than 1000 mutations in the PAH gene, which are listed in a PKU-specific database (PAHvdb in BioPKU [http://www.biopku.org/]). Splicing, nonsense mutations, and out-of-frame insertions or deletions generally result in the loss of PAH (null mutations), while missense mutations and certain in-frame insertion-deletion mutations (indels) cause defective protein translationCitation4. Null mutations in both alleles lead to classic PKU, which is characterized by blood Phe levels >1200 µmol/L if left untreated. These patients benefit from a low Phe diet combined with Phe-free L-amino acid-based protein substitutes, glycomacropeptide (GMP)-based protein substitutes and supplementation with slow-release large neutral amino acids (LNAAs)Citation5,Citation6. This diet prevents rises in blood Phe levels which, according to European guidelinesCitation3, should be continued to maintain blood Phe levels within the 120–360 µmol/L range when <12 years of age and during pregnancy, and in the 120–600 µmol/L range when >12 years of age. Conversely, American guidelines recommend the 120–360 µmol/L target throughout the patient’s lifeCitation7.
Protein substitutes, which also include other micronutrients in many cases, provide essential amino acids and are proven to lower blood Phe levels, probably because they trigger anabolism and alter Phe transport in the intestinal epitheliumCitation3. Even when working towards stricter targets, pediatric patients managed by their families show good compliance to therapy. On the contrary, as reported in a recent Italian multicenter surveyCitation8, adults demonstrate poor compliance with the low Phe nutritional plan (42%). Protein substitutes are also poorly tolerated, as they can cause gastrointestinal discomfort, need to be taken with increased frequency, and generally taste unpleasantCitation3.
PKU is classified according to the needs of the treatmentCitation3. If treated, patients can live a normal life to the point where a considerable percentage of patients (40% in the above-mentioned Italian survey)Citation8 believe that they are not affected by a disease. However, relaxation in diet, which often occurs during the transition from childhood to adolescence, frequently results in missed Phe level targets that result in various symptoms such as attention deficit, insomnia, irritability, mood swings, fatigue, and tremors. European guidelines, therefore, recommend that all adult PKU patients should receive life-long follow-up at specialist metabolic centers, where they should undergo Phe level testing at least monthly, as well as annual outpatient visits depending on their compliance.
This paper presents the results of the first Italian survey exploring the experience of six highly specialized medical centers for the diagnosis and management of PKU, as well as the results of a systematic literature review. The survey was developed to evaluate the management of adult (≥16 years of age) patients with PKU in six Italian centers, with a focus on BH4 treatment, the transition from childhood to adolescence, follow-up, and patient compliance. The overall aim of this study was to analyze and highlight similarities and differences in clinical care between centers, thus providing the basis for a more collaborative approach to maximize clinical management and match unmet needs, where identified.
Methods
Survey
In September 2019, BioMarin Pharmaceutical Inc. (Novato, CA) organized an expert meeting in Bologna, Italy. The expert meeting participants were selected from clinicians across different geographic regions in Italy based on two criteria: (i) known expertise in the field of PKU; and (ii) a high number of adult patients monitored at their PKU reference center.
The following experts were involved: Alberto Burlina (Inherited Metabolic Diseases Division, Regional Center for Expanded Neonatal Screening, Women and Children’s Health Department, University Hospital of Padua, Italy), Maria Teresa Carbone (Pediatric Division, Metabolic and Rare Diseases, Santobono Pausilipon Hospital, Naples, Italy), Vincenzo Leuzzi (Department of Human Neuroscience, Unit of Child Neurology and Psychiatry – University La Sapienza, Rome, Italy), Sabrina Paci (Pediatric Department, ASST Santi Paolo e Carlo, San Paolo Hospital, University of Milan, Milan 20142, Italy), Marco Spada (Department of Pediatrics, Regina Margherita Children’s Hospital, University of Torino, Italy), and Albina Tummolo (Metabolic Diseases Department, Clinical Genetics and Diabetology, Giovanni XXIII Children’s Hospital, Bari, Italy).
The meeting highlighted the need to gain an understanding of the real-life management and follow-up of PKU in adults in Italy. AdRes s.r.l. Health Economics & Outcomes Research (Turin, Italy), in conjunction with BioMarin Pharmaceutical Inc., CD Pharma Group (Milan, Italy), and the expert meeting participants, designed a survey aimed at investigating the following areas associated with PKU: epidemiology, management, healthcare professional involvement, monitoring, BH4 testing, target Phe levels adopted, and pregnancy. A questionnaire (Online Resource 1) was emailed to the expert meeting participants who completed it based on clinical experience at their own center. As some topics were not felt to have been investigated satisfactorily, a second questionnaire was sent out (Online Resource 2), which also explored patient perception, childhood–adulthood transition, and opinions about current and future treatments. Possible conflicting statements/missing data were further investigated in order to collect sufficient data to paint a reliable picture of real-life PKU management in Italy.
All data were analyzed using descriptive statistics and summarized by AdRes s.r.l. Health Economics & Outcomes Research, BioMarin Pharmaceutical Inc., and CD Pharma Group. Results were shared with the expert meeting participants during a teleconference for interpretation and discussion.
Considered issues
In November 2019, BioMarin Pharmaceutical Inc asked AdRes s.r.l. Health Economics & Outcomes Research to conduct a systematic review of literature for inclusion in the reimbursement dossier for the Italian Medicines Agency (AIFA). This work aimed to answer the following clinical questions, formulated by the expert meeting participants in Bologna in September 2019: (i) are PKU patients ≥16 years of age, diagnosed at birth and managed in developed countries, able to maintain blood Phe levels within the recommended range of 600 µmol/L using the low-Phe diet only?; (ii) how does hyperphenylalaninemia affect neurocognitive performance in the adult PKU population?; and (iii) what is the quality of life of the adult PKU population treated with a low-Phe diet alone? The results of the survey and a comparison to the literature are presented in the discussion section of this manuscript.
Systematic review of the literature
The systematic review of the literature involved a search of the PubMed and EMBASE biomedical repositories using keywords. Keywords and controlled terms were used for all fields (author, title, abstract, and text). Search equations were designed using the PICOS method () and developed by means of five search strategies, which all focused on early treated phenylketonuria (ETPKU) in the adult population. The first search strategy was general and focused on “management”, three were specific to scientific issues (“compliance/control”, “neurocognitive outcomes”, and “health-related quality of life”), and the last search strategy was specific to the Italian context (“Italy”). Details of the search terms used are provided in Online Resource 3.
Table 1. PICOS literature search criteria.
The bibliographic search results were imported into the specific collaborative software “Rayyan” (www.rayyan.qcri.org) for the selection phase, in which all the expert meeting participants were involved. After eliminating any duplicates, the articles were initially screened using inclusion and exclusion criteria based on the title and abstract. Full texts were retrieved for articles that were not excluded during this phase, in order to make a final decision about their inclusion. Data extraction and tabulation were then carried out for the included articles. Then, the evidence produced by this review underwent critical analysis and commentary ().
Overall, 878 papers were retrieved; of these, 743 were excluded after analysis of the abstract (103 of these were duplicates). 135 full-text manuscripts were then identified for further review, of which 44 were included in the final review ().
Table 2. Flow of the literature search and selection.
Some studies had overlapping populations and thus it was not possible to quantify the number of patients described exactly. All reports referring to the same study were eliminated, and a most conservative approach for inclusion was applied – that is, if patients were divided into subgroups, only patients in age groups that included patients completely over the age of 16 years were considered. A number range (n) was assigned to the national population described in each study, according to two extreme overlap assumptions: low n value were assigned to national populations with a high number of patients participating in more than one study, meaning a high overlap (NB, only studies with the highest population size for each country was considered for calculation); conversely, the maximum n corresponded to the opposite assumption, that is, no patient had been studied simultaneously in any of the studies included. The resulting number of patients described in the studies was comprised between 3890–5037, across 17 different countries (Australia, Belgium, Canada, Czech Republic, Denmark, Germany, Hungary, Israel, Italy, Netherlands, Norway, Poland, Spain, Switzerland, Turkey, the UK, and the USA).
Results
Literature analysis
The literature review allowed us to outline the global perspective of the clinical management of PKU in adult patients (>16 years).
The analysis revealed the heterogeneity of PKU management in different countries. The greatest variability was found in the definition of PKU phenotypes, target therapeutic levels of blood Phe, and follow-up practices for patients with PKU. In addition, the protocols for BH4 testing varied widely between countries, with sapropterin reported as a treatment option in 34% of respondents in EuropeCitation9.
After a thorough analysis of the literature, the authors focused on three main topics: (i) are PKU patients ≥16 years of age, diagnosed at birth and managed in developed countries, able to maintain blood Phe levels within the recommended range of 600 µmol/L using the low-Phe diet only?; (ii) how does hyperphenylalaninemia affect neurocognitive performance in the adult PKU population?; and (iii) what is the quality of life of the adult PKU population treated with a low-Phe diet alone?
The available evidence indicated that strict adherence to diet therapy is very difficult or impossible in a high percentage of patients (variable, but ∼35–50%); the recommended metabolic control is not achieved, thus resulting in a significant impact on cognitive functions. More than 50% of these patients have blood Phe values >600 µmol/LCitation10–16.
Moreover, prolonged high levels of phenylalaninemia, particularly in adolescence, are associated with a negative impact on the individual’s IQ; regardless of historical metabolic control, hyperphenylalaninemia is associated with acute deficits in cognitive function, such as difficulty concentrating and prolonged reaction times to stimuli, particularly where they involve the need for complex processing. Ultimately, this condition appears to be associated with an increased incidence of psychiatric and behavioral disordersCitation15,Citation17–21.
The burden of the onerous diet therapy, as well as the functional cognitive deficits associated with hyperphenylalaninemia, are the main factors negatively impacting the quality of life as perceived by patients with PKU aged >16 yearsCitation8.
The authors agree on the adoption of European PKU management guidelines as the optimal method to maintain good Phe control and avoid neurocognitive impairment. Accordingly, blood Phe levels recommended in adults are in the 120–600 µmol/L range in patients aged > 12 years, and European Guidelines also recommend that these values are maintained throughout the patient’s life. Moreover, neurocognitive evaluation is recommended at 12 years and 18 years of age in all patients in the presence of poor metabolic control, or if any social or psychological concerns are present (3, statements 10, 11 and 24).
Survey
The total number of PKU patients in Italy is ∼4000 (https://www.osservatoriomalattierare.it/), although a precise estimate is not available because of the lack of a national registry. The six centers participating in the survey declared that they each had between 40 and 320 patients receiving early treatment for PKU, with a total of 678 subjects across all six centers. All patients were aged over 16 years. The majority of PKU patients are currently being monitored and treated; no more than 15% continue to have symptoms ().
Table 3. Adult PKU patients monitoring.
Multidisciplinary management of phenylketonuria
Due to the complex nature of PKU, a multidisciplinary patient approach must be adoptedCitation22.
The six centers participating in this study have an internal team composed of at least one medical specialist, one pediatrician or neuro-pediatrician, and are almost always supported by a dietician and a psychologist ().
Figure 1. Composition of the center’s team.
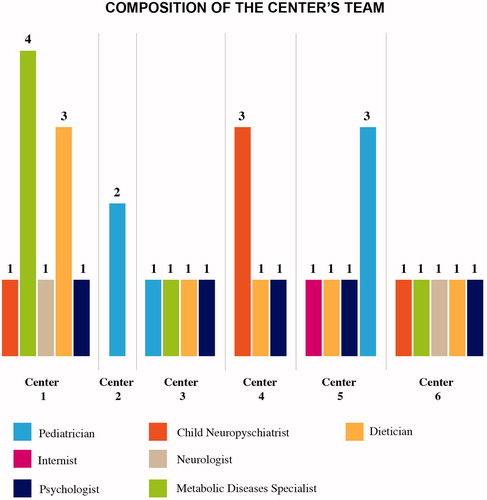
Despite heterogeneities in the composition of the multidisciplinary teams between centers, patient compliance in terms of loyalty to the center and adherence to therapy was not affected. The experts explain that these results are due to the high level of expertise of the specialists, working with PKU patients from the pediatric age to adulthood.
According to all six experts, every adult patient was monitored by the same member of staff during pediatric follow-up, due to active recall policies following the neonatal screening outcome. In adult patients, only rare cases are referred to the centers by a GP or a gynecologist in the event of pregnancy, while some patients decide to report to the center personally.
Treatment
Previous treatment
All centers record and store data relating to the patient’s clinical history. It was therefore possible to trace treatment duration and compliance (). With regard to the patients’ clinical pathway, given the crucial importance of maintaining low phenylalanine levels (in accordance with guidelines) during early life to avoid the development of mental retardation, the experts reported that their patients were on diet therapy up to the age of at least 14 years, with a good level of compliance and a low interruption rate (∼10% between age 10 years and 14 years; ). Diet therapy consisted of replacing all-natural proteins in the diet with safe or phenylalanine-free protein substitutes, consisting of Phe-free L-amino acid-based protein substitutes, glycomacropeptide (GMP)-based protein substitutes and slow-release large neutral amino acids (LNAAs). The diet also contained a balanced intake of all nutrients, energy, vitamins, and minerals. Vitamin and minerals were either added to the protein substitute or given as a separate supplementCitation23.
Table 4. Treatment history of patients under the centers in question.
After age 14 years, a further percentage of subjects (∼20%) stopped treatment, with experts estimating the total abandonment rate to be ∼30% (). All of the experts reported that a portion of patients resumed diet therapy after stopping it in all of the centers ().
Current treatment
The treatment protocol adopted at the six centers requires all patients to receive diet therapy; at one center no treatment was provided to PKU patients with BH4; at the remaining four, the percentage of patients receiving BH4 varied between 2–30% with treatment. It is important to emphasize that one expert always provided continuous support throughout the PKU patient’s treatment. None of the centers used experimental or off-label drugs ().
Table 5. Treatment followed by patients currently under the center’s care.
Compliance
The level of compliance in the adult patient on prescribed treatment was deemed to be low/moderate and not exceeding 65%. As already described in the literature, the experts confirmed that problems adversely affecting compliance mainly concerned the impact of diet therapy on quality of life and particularly the social aspects, as well as a tendency among some patients to deny being affected by a disease. For this reason, three experts believed that the arrival of new therapies could have a positive influence on compliance and on patient willingness to continue follow-up.
Long-term loyalty to the centers was high, with centers generally looking after the same patients throughout their lifetime, from diagnosis to adulthood. The dropout rate after childhood never exceeded 30%. Indeed, the experts argued that, despite poor treatment compliance, patients feared the consequences of total interruption and attend the scheduled annual check-ups. According to the recommendations of European guidelines, patients follow a tighter schedule for visits, with frequency increasing due to treatment changes, social circumstances (such as changing school or leaving home), clinical grounds, and adherence issues (revealed by the monthly Dried Blood Spot (DBS) test follow up)Citation22.
Monitoring
With regard to monitoring the patient’s blood phenylalanine levels, the low phenylalanine range to be maintained is generally that suggested by European guidelines, that is, 120–600 µmol/L (). It is very important to monitor Phe levels constantly. As shown in , each center adopted a different approach to monitoring. For example, Center 5 adopted restricted ranges with no difference between adult and pregnancy doses, while Center 6 adopted a lower high value for pregnancy only, and a higher low value for adults.
Figure 2. Blood Phe range adopted at the centers under analysis. Center 5 adopted restricted ranges with no differences between adult and pregnancy. Center 6 adopted a lower high value for pregnancy only, and a higher low value for adults.
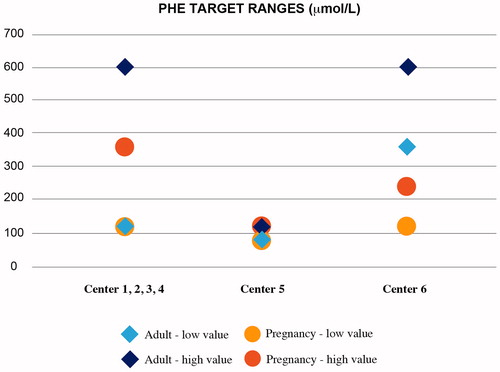
In relation to disease status monitoring (and all the additional assessments that a PKU patient may need), protocols varied significantly among different centers. In fact, phenylalanine levels were measured systematically, with the frequency of such measurements varying from monthly to weekly. Other clinical assessments, such as neuropsychiatric testing, were performed routinely on all patients and on a quarterly basis at one center only. Furthermore, if deemed clinically necessary, magnetic resonance imaging of the brain was also performed. None of the centers routinely carried out a diabetes risk assessment, cardiovascular risk assessment, or lung assessments, as the experts believe these assessments should be carried out according to the patient’s clinical condition and age. The same recommendations regarding comorbidities are stated in the European guidelines (3, statement 20 – grade of recommendation C).
Tetrahydrobiopterin testing
With regard to the methods used for the BH4 responsiveness testCitation24, only one center used the BH4 test for the differential diagnosis to distinguish between PKU/PAH deficiency and PKU/BH4 tetrabiopterin pathway deficiency and did not use this drug in therapy. In this center, BH4 treatment is assigned only to patients suffering from primary BH4 deficiency, while classic and mild PKU patients are treated with a Phe-controlled diet only, following a strict phenylalanine tolerance assessment. In this setting, patients presenting high compliance to diet-therapy maintain blood Phe within the range. Consistently, patients with stable Phe levels <360 µmol/L, on an unrestricted dietary Phe intake, are assigned to the benign phenotype, defined as “HPA no-PKU” – representing the largest category of hyperphenylalaninemic newborns – and no specific treatment is currently indicated for them.
Another center did not test adult patients but performed the loading test on all those with a double mutation not followed by a positive diagnosis at the screening. The remaining four centers retested adult patients who were non-responders as children, and test naïve adults, to evaluate the effectiveness of BH4 as therapeutic support for PKU. The criteria adopted by the five centers when deciding whether to perform the test on a patient differed greatly from center to center. The main test eligibility criterion was the distinctive genetic predisposition of mild/moderate forms. Two centers tested all forms of PKU that do not have a double null mutation, including hyperphenylalaninemia (HPA); one of them tested all neonatal patients; the other three centers tested a reduced number of patients with PKU, with a greater focus on PKU patients affected by mild forms and completely excluding subjects suffering from HPA. Furthermore, the ability to perform the BH4 responsiveness test is not only linked to the patient’s genetic/clinical eligibility, but also to the willingness of the patient’s family to change longstanding eating habits and lifestyle. Some patients also report a real fear of abandoning the diet and refuse to take the test.
The dose of BH4 tested was 20 mg/kg at two centers over a period of 48 h, which was extended to four weeks for unresponsive patients; another center used a 10 mg/kg dose for the first two weeks and 20 mg/kg for the next two weeks, for a total of 4 weeks. One expert stated that they use a dose of 10 mg/kg, which is effective in 90% of cases after two weeks; in non-responders, the dose is increased to 20 mg/kg for two more weeks. At four centers, the percentage of patients who respond to the BH4 test is between 20% and 35%, while for the center that mainly tests mild forms, it reaches up to 70% ().
Figure 3. BH4 responsiveness test execution methods at six centers.

Patient perception
The cognitive difficulties typical of this pathology are often subjectively perceived by patients and are therefore difficult to verify in a standardized and systematic patient assessmentCitation25.
This makes it necessary to carry out a thorough investigation of disease perception among patients with PKU. Some patients report symptoms such as impaired concentration and focus, anxiety, irritability/short temper, memory deficit, headache, and mood swings, all of which are associated with persistent Phe levels above the threshold set by European guidelines (≥600 µmol/L). When Phe levels are within the normal range, patients report improved attention and concentration skills; on the other hand, when the levels are too high patients struggle to recognize their neurocognitive impairment. Only one expert believed that cognitive dysfunction could somehow affect follow-up.
Transition from childhood to adulthood
The experts argued that the patients’ transition process needs to be implemented, providing a gradual switch to the management of the adult patient by a new physician responsible for the patient during and after the transition, in order to follow his/her requirements as an adult. Currently, this type of professional figure is missing in all of the six centers. Moreover, the experts indicated that patients and their families are reluctant to change the clinicians who have monitored them for years. A structured transition process appeared to be successful for most of the patients surveyed in a German study, with a strong increase in patient satisfaction both from a psychosocial and socioeconomic point of viewCitation26.
Data show that effective PKU patient transitions to a specific center for adults are limited by the lack of metabolic expertise, which is currently largely concentrated in pediatric centers. As suggested by one expert and agreed by the others, greater primary care involvement would be beneficial in the management of patients with PKU, both to facilitate the transition process and to manage specific aspects of the disease and its comorbidities. There was a suggestion to create a network of preferential communication channel, with the aim of the patients obtaining total comprehensive support. A professional figure taking care of adult PKU patients could also be crucial in recovering subjects who are lost to follow-up during the transition.
Lastly, it is interesting to note that treatment compliance and ongoing follow-up exceeded 14% in subjects treated with diet therapy and BH4 during this delicate phase (). In fact, as these patients have better clinical measurements and are encouraged to carry out the tests, they seem to be more responsible and motivated than those forced to follow a more stringent diet, especially in cases where a diet supported by BH4 allows them to obtain a real increase in phenylalanine tolerance, consequently leading to a partial or total reduction of their dietary restrictions.
The potential reasons for the loss of motivation during the transition have been summarized by the experts as follows: the move away from parental control; the refusal to comply with a treatment that has such a big impact on their social and personal life; diet-related logistical and organizational difficulties; the embarrassment of admitting to health professionals that they have failed to comply with therapeutic instructions; poor perception of their disease; and even the denial of their diagnosis.
Pregnancy
An adequate level of alignment with a good and homogeneous rate of accuracy in maternal PKU follow-up was found in all centers. The Phe ranges for pregnant women were mostly 120–360 µmol/L, as recommended by European guidelines (). Phe level monitoring was more frequent than in non-pregnant individuals. Three experts stated that the DBS must be submitted once to twice weekly, one required them to be submitted every two days, while the other two followed European guidelinesCitation3; however, the experts agreed that pregnancy requires close attention to adapting both the treatment and monitoring to the specific requirements of the patientCitation27.
According to three experts, the diet must be accurately balanced to ensure an adequate calorie and protein intake that varies according to the gestational period and depending on the presence of nausea and vomiting and the increase in Phe tolerance that physiologically occurs during pregnancy.
Beyond the specific indications provided by each center for the optimal management of these patients, it is appropriate to focus on the risks connected with pregnancy in women with PKU, although this awareness is present in almost all cases (). Experts agree that all patients should return to their PKU management center in the event of pregnancy, regardless of their treatment or Phe levels. According to one expert, this return is also closely related to blood Phe concentrations above the threshold values.
Figure 4. PKU during pregnancy, awareness, and gynecological management.

One expert highlighted the importance of the specific monitoring required after pregnancy, which implies a need for comprehensive follow-up of the mother during breastfeeding and monitoring of the baby’s general state of health until at least 6 years of age.
Future strategies
Another result that emerged from the surveys related to the experts’ opinion of the strategies currently available in Italy (diet and BH4, combined or separately) and the possible impact of future strategies on the treatment of PKU patientsCitation28.
At present, dietary management alone may not be sufficient to treat adult patients, while BH4 and dietary management is a valid therapeutic approach for responsive patients. It has been emphasized that optimal treatment with diet therapy is even more complicated to achieve if current levels of compliance among adult patients are considered (see ), the reduced perception of a patient’s own disease that impacts compliance and, in the case of BH4, the greatest difficulty is in relation to the need for systematic retesting in adult patients.
With regard to emerging therapies and the impact they could have on the lifelong management of patients with PKU, they will certainly be embedded in the new standards of PKU care for patients unable to comply with diet therapy and maintain their phenylalanine levels below the threshold values. In any case, there is mutual agreement on the possibility of allowing a completely free diet where risks do not exceed benefits and where effectiveness is proved by blood Phe levels remaining within the target range.
Discussion
To thoroughly investigate the real-life contemporary management of PKU in Italy, six Italian experts conducted a systematic review of the literature on selected international publications and administered a survey to better understand some relevant clinical aspects. The expert panel has managed a total of 678 patients aged over 16 years with early treated PKU across their six centers.
Considering the results of the systematic review of the literature, the survey results, and the experts’ personal experience, several findings have been discovered and discussed during meetings. The main topics have been the composition of the multidisciplinary team, Phe target ranges in centers, therapeutic strategies, follow-up schedules, and expectations regarding future treatments.
Our survey highlighted that patients are mostly managed by the pediatric reference center, from the initial diagnosis of PKU during the newborn screening through to adulthood without any transition to a specialized adult clinician. As already shown in a survey based on co-creative sessions involving PKU patients in Italy, the continuity of follow-up allows clinicians to closely monitor the patients and track their compliance with therapy throughout their lives. In addition, the perception of doctor–patient communication and relationship with the healthcare system is the main factor that facilitates adherence and engagement in the management of PKU. However, a profound revision of the PKU care system in Italy is strongly desired with particular attention to the transition phase, a critical step for patient complianceCitation29.
Adherence and compliance are low, due to the impact on the quality of life, social limitations, and self-denial that patients have PKUCitation8. Patients followed-up in the selected centers are totally on a restricted diet supplemented with protein substitutes and Phe-free formulas in 95% of cases. No more than 30% of the total treated patients use BH4 to take advantage of the reduced dietary restrictions, improved food tolerance, and increased daily protein intake allowance. For these reasons, adherence and compliance are higher in BH4-treated patients.
Despite assimilating the indications given in the European guidelinesCitation3, our six centers adopt different strategies in many contexts. For example, in the selection and testing of patients who can be treated with BH4, there are differences among centers with regard to both the time of testing and the dosages scheduled. The treatment trial in potential BH4-responders may occur over a few weeks or even monthsCitation3. One center described 24 h BH4 loading testing using 20 mg/kg to differentiate between BH4-responders and non-responders. In the others, also with different times and dosages, the 48-h test followed by a 4-week test appears to be useful to detect slow responders as well as responsiveness in more severe phenotypes.
It should be highlighted that the possibility of performing the BH4 responsiveness testCitation30 is associated with the patient’s genetic/clinical eligibility, with clinicians preferring mild/moderate phenotypes, and also with the willingness of the patient’s family to modify their consolidated dietary habits and lifestyles.
Following this evidence there are many difficulties in achieving target ranges in many cases, that indeed are the same as those used across most centers as required by European guidelines.
A heterogeneous follow-up among the centers was found for all that concerns DBS frequency and Phe level monitoring, from weekly up to monthly, as well as neuropsychiatric evaluations being administrated quarterly in only one center. Endocrinological, respiratory, and cardiovascular examinations were performed only in selected cases.
Noteworthies are the patient disease perceptions and neurocognitive considerations. The survey showed that fogginess, concentration reduction, low attention, anxiety, irritability, memory deficit, headache, and unstable mood are common features in a patient with uncontrolled Phe (generally above 600 µmol/L). These symptoms can be unperceived but then recognized when Phe levels are close to physiological levelsCitation17.
The final observations can be summarized as follows:
There are differences in PKU management
Diet therapy patient compliance in Italy is suboptimal
Diet therapy alone may not be sufficient to treat adult patients
BH4 therapeutic approaches are used to various degrees
The BH4 responsiveness test is performed in different ways
Patients perception of their own disease is low
Conclusions
In conclusion, we have reinforced the importance of a homogeneous and shared approach to PKU patients, describing the Italian real-life clinical experience of managing PKU in order to encourage and implement therapeutic strategies and follow-up, with the aim to increase patients compliance and adherence as well as achieving Phe levels in line with the European guidelines recommended ranges. Correct and standardized use of therapeutic options available to date is highly recommended to be implemented in all centers. The follow-up of adults and the identification of their neurocognitive and comorbidity needs is recommended, as well as focus on their self-awareness regarding their disease together with an implementation of the transition process with major involvement of primary care.
Although time and effort will be required, not only by the surveyed experts but at a national level, we highly recommend the creation of a network between centers and territories. The network will offer the opportunity to share experiences, be aligned and manage Italian patients homogeneously, and create the basis for effectively improving the lives of patients with PKU in Italy.
Transparency
Declaration of funding
This work was supported by BioMarin Pharmaceutical Inc.
Declaration of financial/other relationships
AB has received advisory board honoraria, speaker fees, and travel support from BioMarin, Nutricia, PIAM Farmaceutici, Sanofi Genzyme, Takeda Shire, and Orphan Recordati. VL has received advisory board honoraria, speaker fees, and travel support from Nutricia, BioMarin, PTC, and PIAM Farmaceutici. MS has received, in the last two years, advisory board honoraria, speaker fees, and travel support from Sanofi, PIAM Farmaceutici, Takeda Shire, BioMarin, Nutricia, Sobi, Amicus, APR, and Orphan Recordati. MTC has received advisory board honoraria and travel support from BioMarin, PIAM Farmaceutici, APR, and Vitaflo. SP has received funding from BioMarin, Nutricia, APR, Mamoxi, PIAM, and Orphan Recordati. AT was an invited advisor for BioMarin, Nutricia, Recordati, and APR advisory boards and received project funding from BioMarin. Peer reviewers on this manuscript have received an honorarium from CMRO for their review work. One reviewer is an advisory board member for Merck Serono, Biomarin, Applied Pharma Research, and Nutricia and has received speaker’s fees from Applied Pharma. Another reviewer has participated in research for Merck Serono, BioMarin, Nutricia, Vitaflo, Cambrooke, PIAM, and Lifediet and currently works for Danone, the owner of Nutricia, which produces specialized nutrition products including those intended for the dietary management of PKU. The remaining reviewers have no relevant financial relationships or otherwise to disclose.
Author contributions
All authors have made substantial intellectual contributions to the conception of the survey and the analysis and interpretation of the data. They have all been involved in drafting the manuscript and revising it critically for important intellectual content.
Supplemental Material
Download MS Word (62.7 KB)Acknowledgements
The authors thank AdRes s.r.l. Health Economics & Outcomes Research who managed the data collection. They acknowledge Cristoforo Incorvaia, senior medical writer, and CD Pharma Group, who provided medical writing services on behalf of BioMarin Pharmaceutical Inc.
References
- Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376(9750):1417–1427.
- Muntau AC, Adams DJ, Bélanger-Quintana A, et al. International best practice for the evaluation of responsiveness to sapropterin dihydrochloride in patients with phenylketonuria. Mol Genet Metab. 2019;127(1):1–11.
- van Wegberg AMJ, MacDonald A, Ahring K, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis. 2017;12(1):162.
- Danecka MK, Woidy M, Zschocke J, et al. Mapping the functional landscape of frequent phenylalanine hydroxylase (PAH) genotypes promotes personalised medicine in phenylketonuria. J Med Genet. 2015;52(3):175–185.
- MacLeod EL, Ney DM. Nutritional management of phenylketonuria. Ann Nestle Eng. 2010;68(2):58–69.
- Burlina AP, Cazzorla C, Massa P, et al. The impact of a slow-release large neutral amino acids supplement on treatment adherence in adult patients with phenylketonuria. Nutrients. 2020;12(7):2078.
- Vockley J, Andersson HC, Antshel KM, et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med. 2014;16(2):188–200.
- Cazzorla C, Bensi G, Biasucci G, et al. Living with phenylketonuria in adulthood: the PKU ATTITUDE study. Mol Genet Metab Rep. 2018;16:39–45.
- Blau N, Belanger-Quintana A, Demirkol M, et al. Management of phenylketonuria in Europe: survey results from 19 countries. Mol Genet Metab. 2010;99(2):109–115.
- Pilotto A, Blau N, Leks E, et al. Cerebrospinal fluid biogenic amines depletion and brain atrophy in adult patients with phenylketonuria. J Inherit Metab Dis. 2019;42(3):398–406.
- Jurecki ER, Cederbaum S, Kopesky J, et al. Adherence to clinic recommendations among patients with phenylketonuria in the United States. Mol Genet Metab. 2017;120(3):190–197.
- Crujeiras V, Aldámiz-Echevarría L, Dalmau J, et al. Vitamin and mineral status in patients with hyperphenylalaninemia. Mol Genet Metab. 2015;115(4):145–150.
- Procházková D, Jarkovský J, Vinohradská H, et al. Controlled diet in phenylketonuria and hyperphenylalaninemia may cause serum selenium deficiency in adult patients: the Czech experience. Biol Trace Elem Res. 2013;154(2):178–184.
- MacDonald A, Nanuwa K, Parkes L, et al. Retrospective, observational data collection of the treatment of phenylketonuria in the UK, and associated clinical and health outcomes. Curr Med Res Opin. 2011;27(6):1211–1222.
- Vilaseca MA, Lambruschini N, Gómez-López L, et al. Quality of dietary control in phenylketonuric patients and its relationship with general intelligence. Nutr Hosp. 2010;25(1):60–66.
- Gokmen-Ozel H, MacDonald A, Daly A, et al. Long-term efficacy of ‘ready-to-drink’ protein substitute in phenylketonuria. J Hum Nutr Diet. 2009;22(5):422–427.
- Romani C, Palermo L, MacDonald A, et al. The impact of phenylalanine levels on cognitive outcomes in adults with phenylketonuria: effects across tasks and developmental stages. Neuropsychology. 2017;31(3):242–254.
- Pietz J, Landwehr R, Kutscha A, et al. Effect of high-dose tyrosine supplementation on brain function in adults with phenylketonuria. J Pediatr. 1995;127(6):936–943.
- Pietz J, Dunckelmann R, Rupp A, et al. Neurological outcome in adult patients with early-treated phenylketonuria. Eur J Pediatr. 1998;157(10):824–830.
- Huijbregts SCJ, Bosch AM, Simons QA, et al. The impact of metabolic control and tetrahydrobiopterin treatment on health related quality of life of patients with early-treated phenylketonuria: a PKU-COBESO study. Mol Genet Metab. 2018;125(1–2):96–103.
- Camfield CS, Joseph M, Hurley T, et al. Optimal management of phenylketonuria: a centralized expert team is more successful than a decentralized model of care. J Pediatr. 2004;145(1):53–57.
- van Spronsen FJ, van Wegberg AM, Ahring K, et al. Key European guidelines for the diagnosis and management of patients with phenylketonuria. Lancet Diabetes Endocrinol. 2017;5(9):743–756.
- Rocha JC, MacDonald A. Dietary intervention in the management of phenylketonuria: current perspectives. Pediatric Health Med Ther. 2016;7:155–163.
- Blau N, Hennermann JB, Langenbeck U, et al. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab. 2011;104:S2–S9.
- Jahja R, van Spronsen FJ, de Sonneville LMJ, et al. Social-cognitive functioning and social skills in patients with early treated phenylketonuria: a PKU-COBESO study. J Inherit Metab Dis. 2016;39(3):355–362.
- Mütze U, Roth A, Weigel JFW, et al. Transition of young adults with phenylketonuria from pediatric to adult care. J Inherit Metab Dis. 2011;34(3):701–709.
- Prick BW, Hop WCJ, Duvekot JJ. Maternal phenylketonuria and hyperphenylalaninemia in pregnancy: pregnancy complications and neonatal sequelae in untreated and treated pregnancies. Am J Clin Nutr. 2012;95(2):374–382.
- Strisciuglio P, Concolino D. New strategies for the treatment of phenylketonuria (PKU). Metabolites. 2014;4(4):1007–1017.
- Borghi L, Moreschi C, Toscano A, et al. The PKU & ME study: a qualitative exploration, through co-creative sessions, of attitudes and experience of the disease among adults with phenylketonuria in Italy. Mol Genet Metab Rep. 2020;23:100585.
- Zori R, Ahring K, Burton B, et al. Long-term comparative effectiveness of pegvaliase versus standard of care comparators in adults with phenylketonuria. Mol Genet Metab. 2019;128(1–2):92–101.