
Abstract
Adenylosuccinic acid (ASA) modifies Duchenne muscular dystrophy (DMD) progression in dystrophic mdx mice and human DMD patients. Despite an established role for ASA in augmenting metabolism and cellular energy homeostasis, our previous data suggests an undiscovered ulterior mode of action capable of modifying DMD disease course. Here, we identify ASA as a novel inducer of nuclear factor erythroid 2-related factor-2 (Nrf2), master regulator of the antioxidant and cytoprotective response to cell stress.
Duchenne Muscular Dystrophy (DMD), an incurable neuromuscular disorder, is fatal in all cases due to progressive skeletal and cardiac muscle wasting in miceCitation1 and humansCitation2. Standard-of-care corticosteroid treatment has prevailed for two decades and, combined with nocturnal ventilation, has increased the lifespan of amenable DMD patients significantlyCitation3. However, childhood loss of ambulatory function has persisted as a relatively unmodifiable prognostic event which severely impacts patient (and family) quality of life. Recently approved gene therapies represent promising progress in therapeutic development despite efficacy and long-term safety being equivocalCitation3. With high unmet clinical need, the hunt for efficacious therapeutics that improve both patient quality and quantity of life is crucial.
In clinical trials conducted by the late Dr. Charles Bonsett (1980–1990s), adenylosuccinic acid (ASA) was identified as a candidate therapeutic with strong translational potential as it attenuated DMD progressionCitation2. ASA is made endogenously by ASA synthetase from inosine in the purine nucleotide cycle (PNC), which is augmented during metabolic stress to recover degraded purines and stimulate the mitochondria to re-balance energy homeostasisCitation4. Since DMD was considered a metabolic disease during that era (i.e. pre-discovery of DMD’s genetic origin), ASA’s mode of action (MOA) was attributed to its metabolic activity. Due to ASA’s high production cost, infusion difficulties, and the eventual inability to source sufficient quantities for long-term treatment, these trials were discontinued despite clinical data suggesting persistent efficacy long after treatment cessationCitation2.
Recently, we confirmed that ASA can also attenuate murine (mdx) DMDCitation1. Despite profound benefits on skeletal muscle histopathology, we saw only modest effects of ASA on metabolic and mitochondrial function parameters. These data suggested to us an ulterior MOA for ASA aside from metabolic regulation, which appears more influential on mitigating the severity and progression of DMD. We suspected activation of nuclear factor erythroid-2 related factor-2 (Nrf2) since ASA: (1) attenuated the superoxide content of DMD myoblastsCitation1 and; (2) is a fumarate-generating compound and pharmacological fumarate donors, such as dimethyl fumarate (DMF), are well-established Nrf2 activatorsCitation5. Herein, we assessed Nrf2 protein expression in skeletal muscles from our previous study to confirm whether induction of this master cytoprotective regulator was the putative MOA. Nrf2 was quantified by immunoblotting (rabbit, 1:1000, Gene Signaling, #12721) in quadriceps muscles from 12-week-old male C57Bl/10ScSn (healthy CON) and C57Bl/10mdx (dystrophic mdx) mice treated with/without ASA (3-3000 µg/mL) for 8-weeksCitation1.
Nrf2 is an important facilitator of hormesis: the adaptive response to cellular stress which promotes survival. Endogenously, Nrf2 elicits a graded cytoprotective response proportional to the scale and repetition of cytotoxic insult. Thus, basal Nrf2 expression reflects the current cellular stress climate. Basal Nrf2 expression was comparable between CON and mdx quadriceps consistent with stablised disease in mdx mice of this age (). The mdx mouse recapitulates a milder disease course and disease stabilisation from ∼1 to 12 m age. In contrast, oxidative stress, muscle damage and inflammation is rampant in skeletal muscles from juvenile (0–1.5 m) and aged (>12 m) mice. Basal Nrf2 expression patterns are currently uncharacterised in severe/progressive murine DMD.
Figure 1. Adenylosuccinic Acid (ASA) is a novel inducer of nuclear factor erythroid 2-related factor 2 (Nrf2). Basal Nrf2 protein expression is comparable but ASA treatment induces Nrf2 in CON and mdx quadriceps (p < .05 treatment effect (A). Data is mean Nrf2 protein normalized to the total protein signal in in each lane ± SEM with representative bands; n = 6–8. In a proposed mechanism of action (B), ASA is converted to bioactive fumarate (FUM) either in the gut epithelium or in target tissues which express adenylosuccinate lyase (ASL). ASL converts ASA to FUM and recovers inosine monophosphate (IMP) to adenosine monophosphate (AMP) in the purine nucleotide cycle (PNC). ASA is endogenously re-synthesized through adenylosuccinate synthase (ASS). FUM is shuttled into the mitochondria to stimulate oxidative phosphorylation or binds cytosolic Kelch-like ECH-associated protein-1 (Keap1) to interrupt Nrf2 repression. This prevents Nrf2 ubiquitination and degradation leading to protein accumulation (A&B). Nrf2 translocates nuclearly where it heterodimerizes with MAF proteins to transcribe the antioxidant response element (ARE) and induce the cytoprotective and antioxidant response. Circulating FUM can also activate the hydroxycarboxylic acid receptor 2 (HCAR2) which, like Nrf2, exerts strong anti-inflammatory activity through the inhibition of nuclear factor kappa B (NF-κB). Created with BioRender.com.
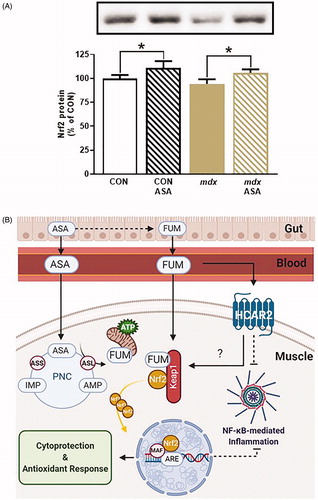
In DMD patients, Nrf2 expression and activation paradoxically declines with age as weight-bearing activity and disease severity escalatesCitation6. While the mechansisms concerning this apparent failure of hormesis are unclear, a similar scenario is portrayed in sarcopenic muscle wastingCitation7. In aged skeletal muscle at least, supressed Nrf2 expression/activity are amenable to exercise stimuliCitation7 suggesting that pharmacological/metabolic Nrf2 targeting could be beneficial to re-establish hormetic cytoprotection in DMD muscles. Our data demonstrate that ASA potentiates Nrf2 expression in both healthy and dystrophic muscles (). Nrf2 is negatively regulated by Kelch-like ECH-associated protein-1 (Keap1) which, under basal conditions, binds cytosolic Nrf2 stimulating its degradationCitation5 (). Chemical modification of Keap1 by inducers (i.e. fumarate, endogenous free radicalsCitation5) precludes Keap1-dependent Nrf2 degradation causing cytosolic Nrf2 accumulation. Thus, increased Nrf2 protein content is synonomous with Keap1 cleavage and Nrf2 activation. Our new data highlight ASA as a novel inducer of Nrf2 in this regard and explain the efficacy we have described previouslyCitation1.
Following dissociation from Keap1, Nrf2 translocates nuclearly where it binds the antioxidant response element (ARE) enhancer, initiating transcription of downstream target genes and the antioxidant and cytoprotective responseCitation5 (). Nrf2 also modulates the transcription of mitochondrial biogenesis genes (i.e. mitochondrial transcription factor A (TFAM))Citation8, and thus directly regulates mitochondrial pool maintenance. These Nrf2-dependent modifications reflect our published data in ASA-treated myoblasts derived from DMD and healthy patients, in which superoxide levels diminished and mitochondrial content increasedCitation1. Our data are consistent with the known cytoprotective effects of Nrf2 activation.
ASA likely induces Nrf2 through driving PNC-mediated synthesis of endogenous fumarate (). This MOA assumes that ASA is bioavailable and able to penetrate the sarcolemma. That ASA treatment decreases adenosine monophosphate-activated protein kinase (AMPK) phosphorylation in healthy muscles suggest both bioavailability and PNC integration sufficient to re-balance energy levels (i.e. increase the ATP/AMP ratio)Citation1. Alternatively, ASA may be converted to fumarate in gut epithelium and circulated in this bioactive form. Fumarate donors are well-established Nrf2 activators through multiple modalities. Monomethyl fumarate, the bioactive form of the approved Multiple Sclerosis drug, DMF, induces Nrf2 both directly through fumarate modulation of Keap1 and indirectly through the hydroxycarboxylic acid receptor 2 (HCAR2)Citation5. Both pathways exert potent anti-inflammatory effects through nuclear factor kappa B (NF-κB) inhibitionCitation5. Anti-inflammation could be highly influential in ASA-dependent modification of the severity and progression of human and murine DMDCitation1,Citation2.
Here, we highlight ASA as a novel inducer of the Nrf2-mediated cytoprotective response through long-term supplementation, and Nrf2 as a feasible drug target to treat DMD. Further studies, e.g. using luciferase-based reporters of ARE transcription, will confirm the downstream antioxidant/cytoprotective response elicited by ASA.
Transparency
Declaration of funding
This paper was not funded.
Declaration of financial/other relationships
ER is a consultant to Santhera Pharmaceuticals. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgements
None stated.
References
- Timpani CA, Goodman CA, Stathis CG, et al. Adenylosuccinic acid therapy ameliorates murine Duchenne Muscular Dystrophy. Sci Rep. 2020;10(1):1–18.
- Bonsett C, Rudman A. The dystrophin connection—ATP? Med Hypotheses. 1992;38(2):139–154.
- Agboola F, Lin GA, Fluetsch N, et al. The effectiveness and value of Deflazacort and Exon-Skipping therapies for the management of Duchenne muscular dystrophy: a summary from the Institute for Clinical and Economic Review’s New England Comparative Effectiveness Public Advisory Council. J Manag Care Spec Pharm. 2020;26(4):361–366.
- Timpani CA, Hayes A, Rybalka E. Revisiting the dystrophin–ATP connection: how half a century of research still implicates mitochondrial dysfunction in Duchenne muscular dystrophy aetiology. Med Hypotheses. 2015;85(6):1021–1033.
- Kourakis S, Timpani CA, de Haan JB, et al. Targeting Nrf2 for the treatment of Duchenne Muscular Dystrophy. Redox Biol. 2021;38:101803.
- Petrillo S, Pelosi L, Piemonte F, et al. Oxidative stress in Duchenne muscular dystrophy: focus on the NRF2 redox pathway. Hum Mol Genet. 2017;26(14):2781–2790.
- Safdar A, deBeer J, Tarnopolsky MA. Dysfunctional Nrf2-Keap1 redox signaling in skeletal muscle of the sedentary old. Free Radic Biol Med. 2010;49(10):1487–1493.
- Huang D-D, Fan S-D, Chen X-Y, et al. Nrf2 deficiency exacerbates frailty and sarcopenia by impairing skeletal muscle mitochondrial biogenesis and dynamics in an age-dependent manner. Exp Gerontol. 2019;119:61–73.